Identification of Pre-Diagnostic Metabolic Patterns for Glioma Using Subset Analysis of Matched Repeated Time Points



Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Progression Pattern Analysis
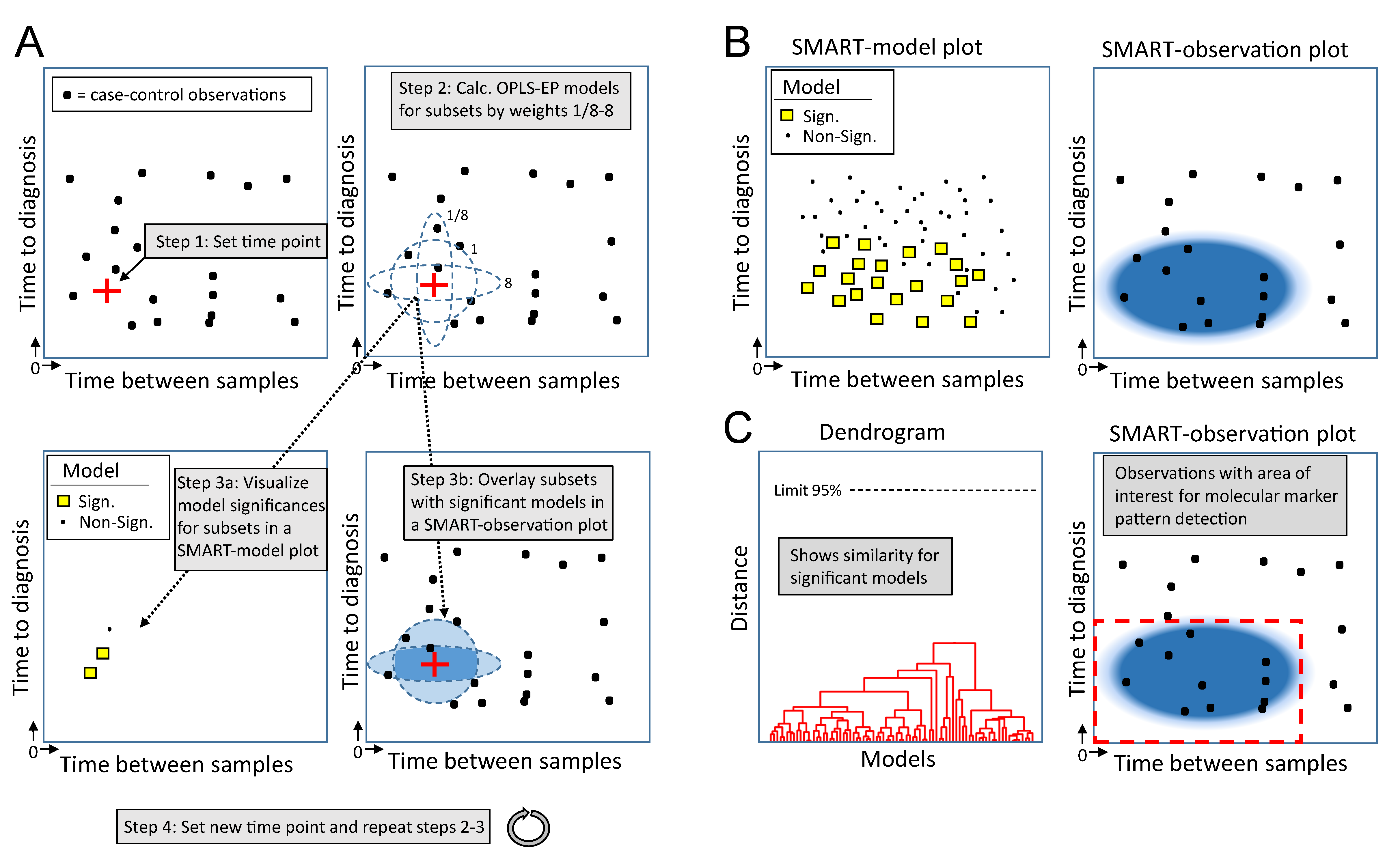
2.2. Outline of the SMART Procedure
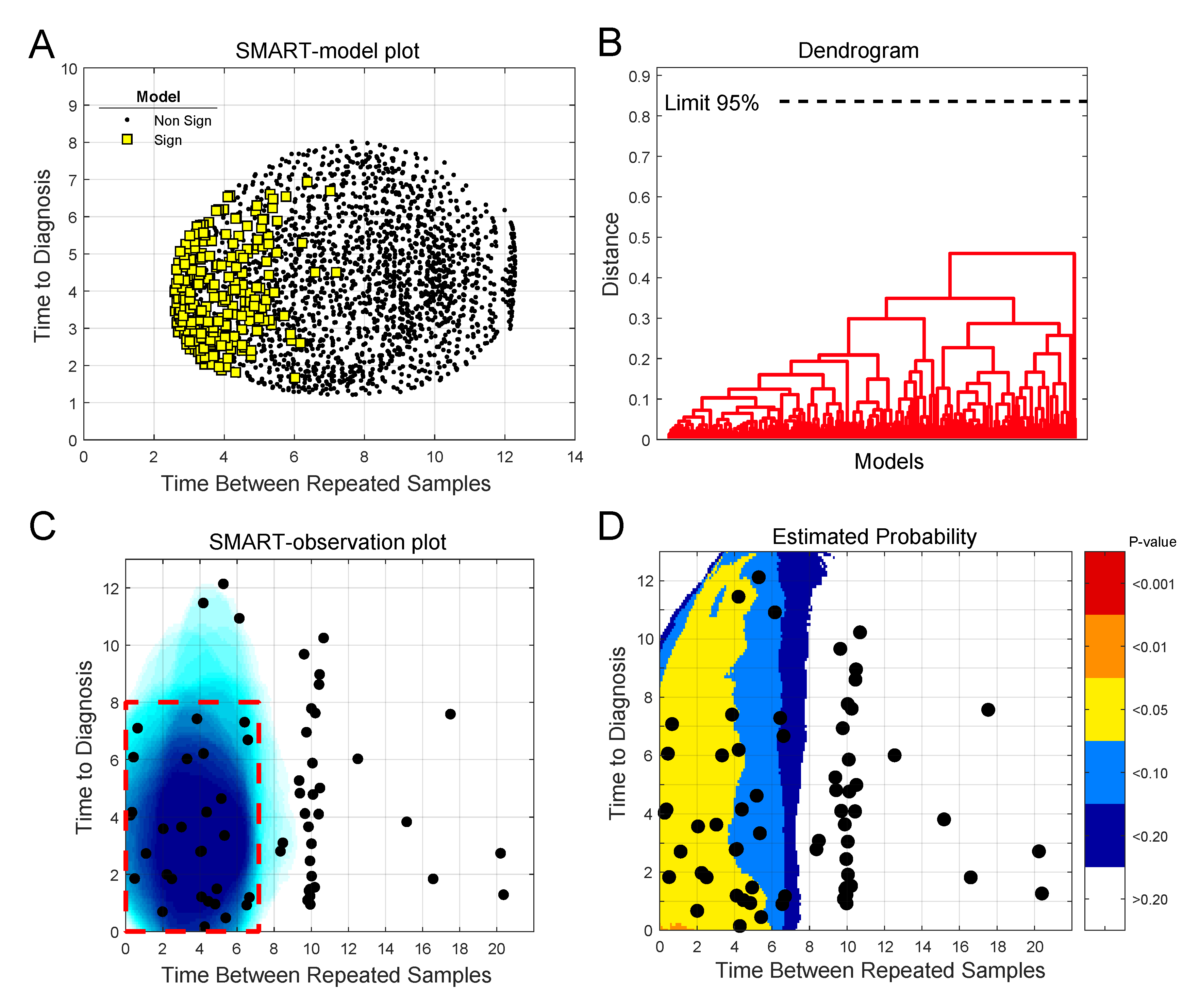
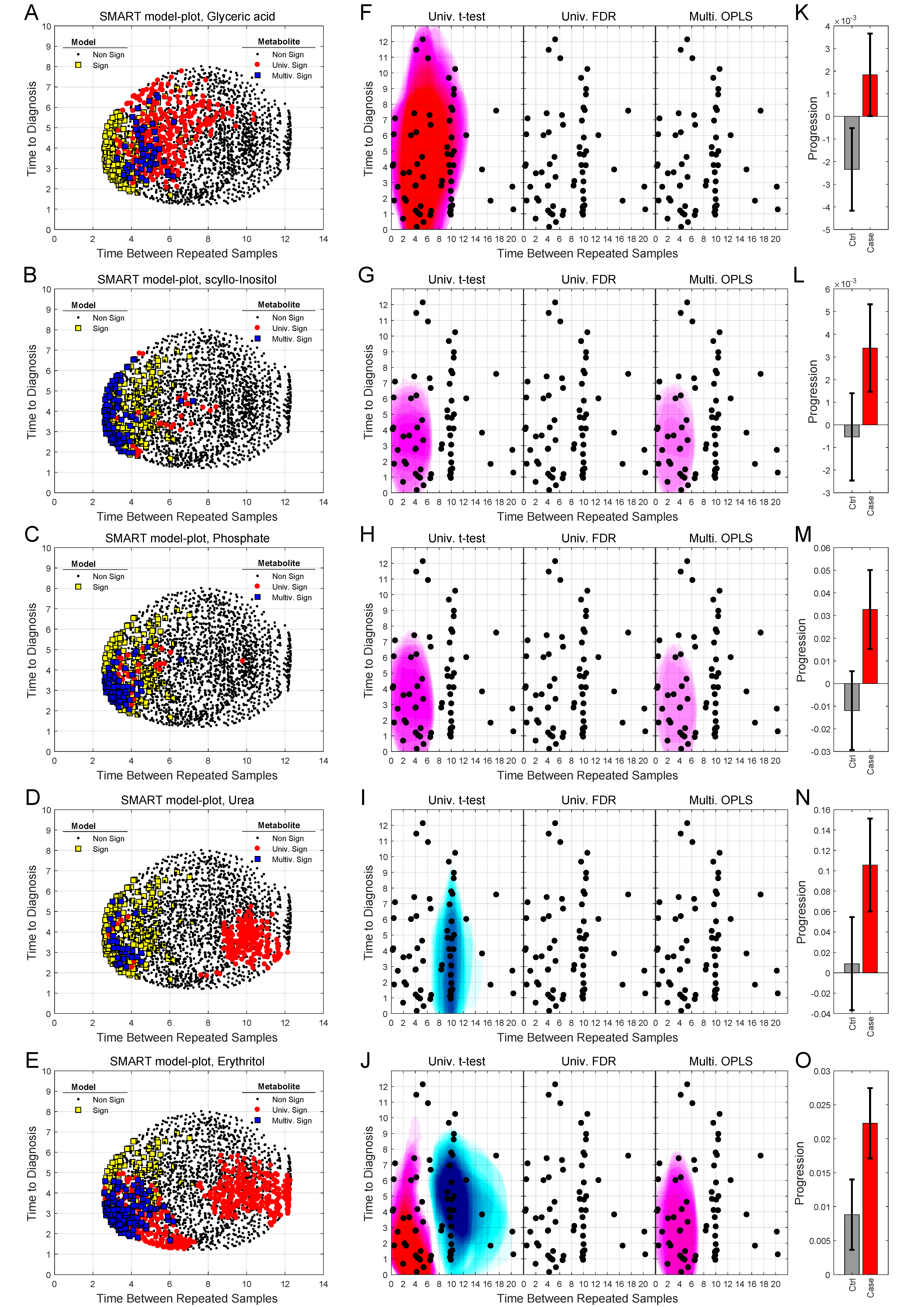
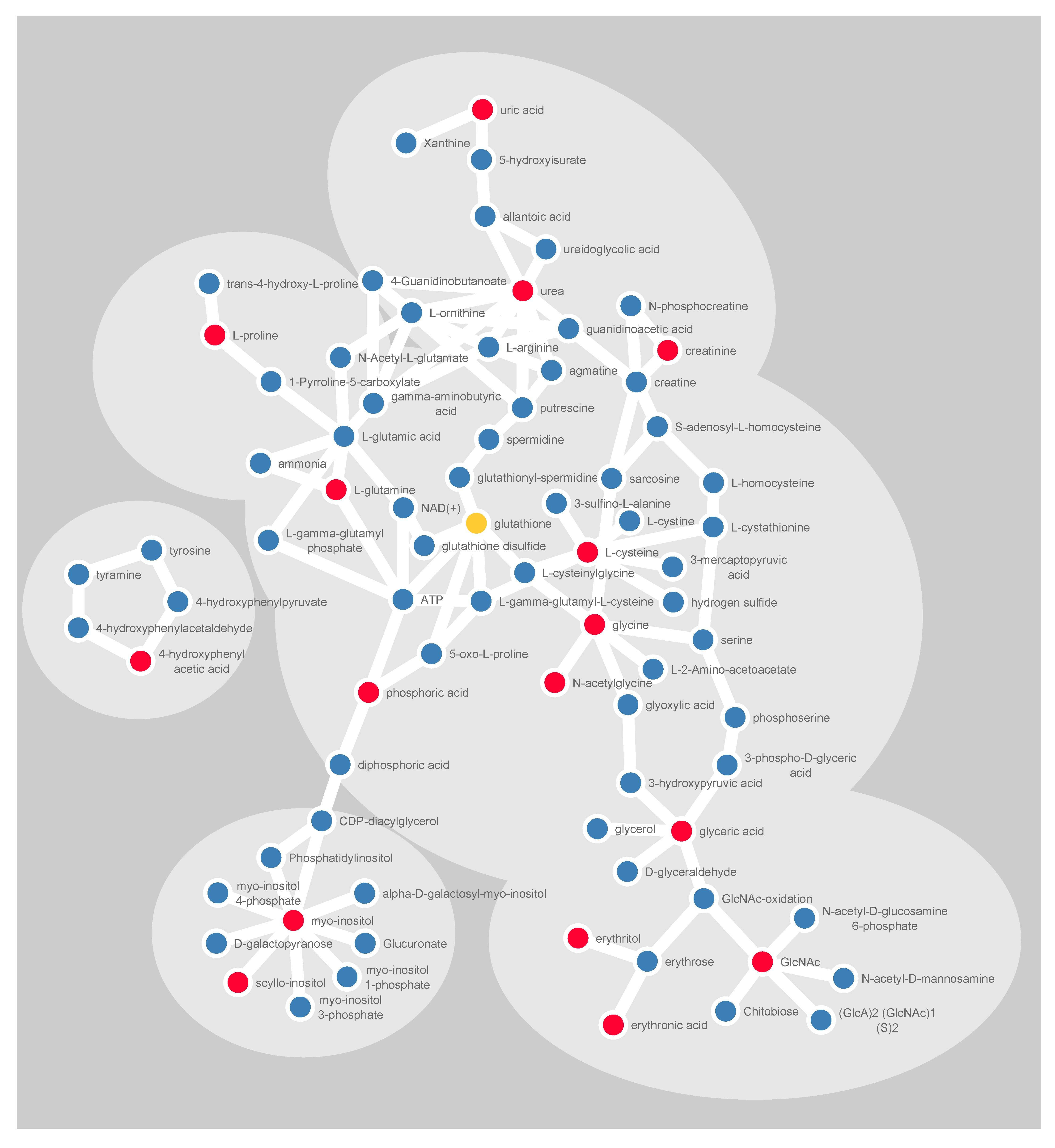
2.3. Detection of Early Metabolic Marker Patterns for Glioma
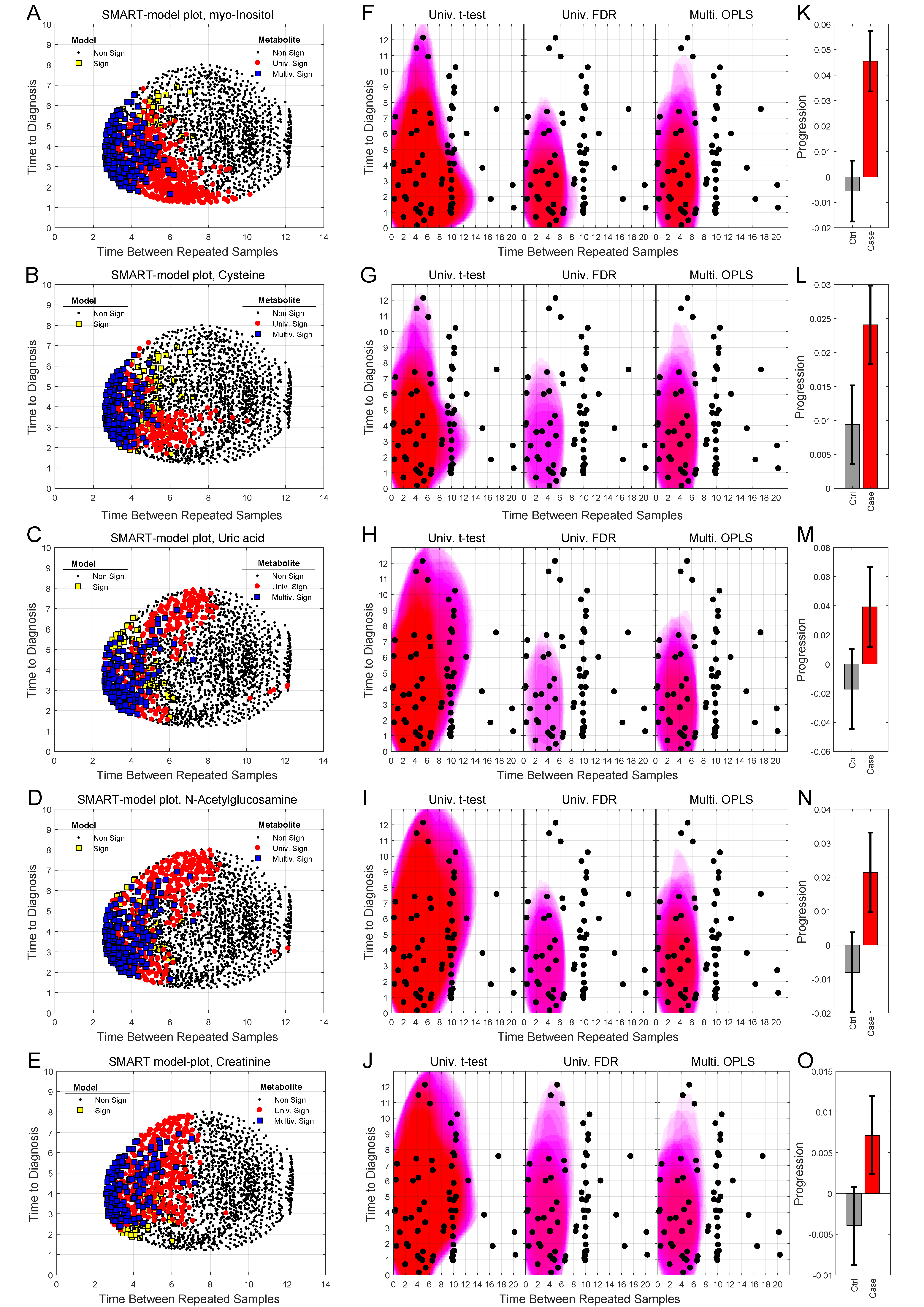
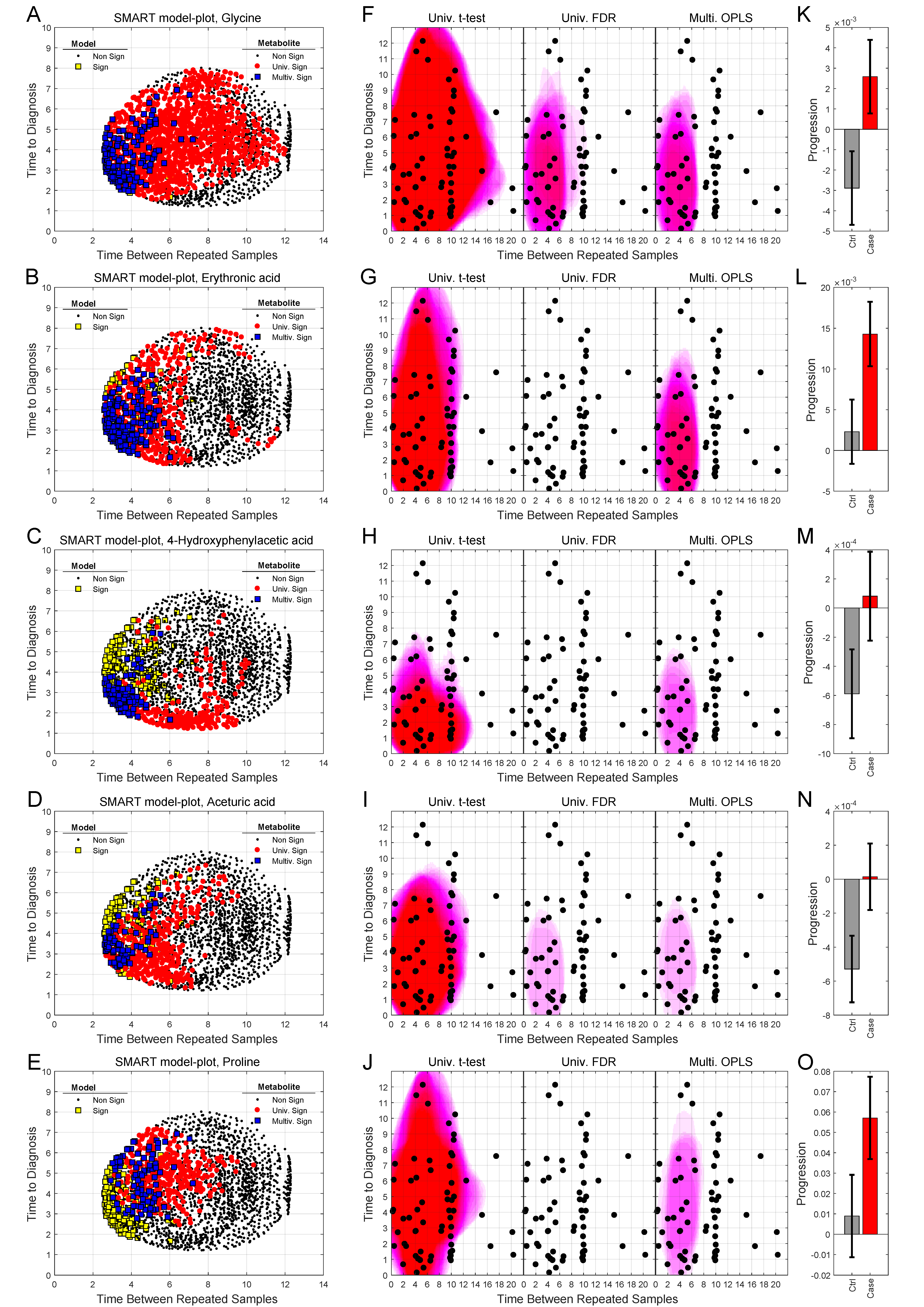
2.4. Analysis of Progression Patterns for Individual Metabolites
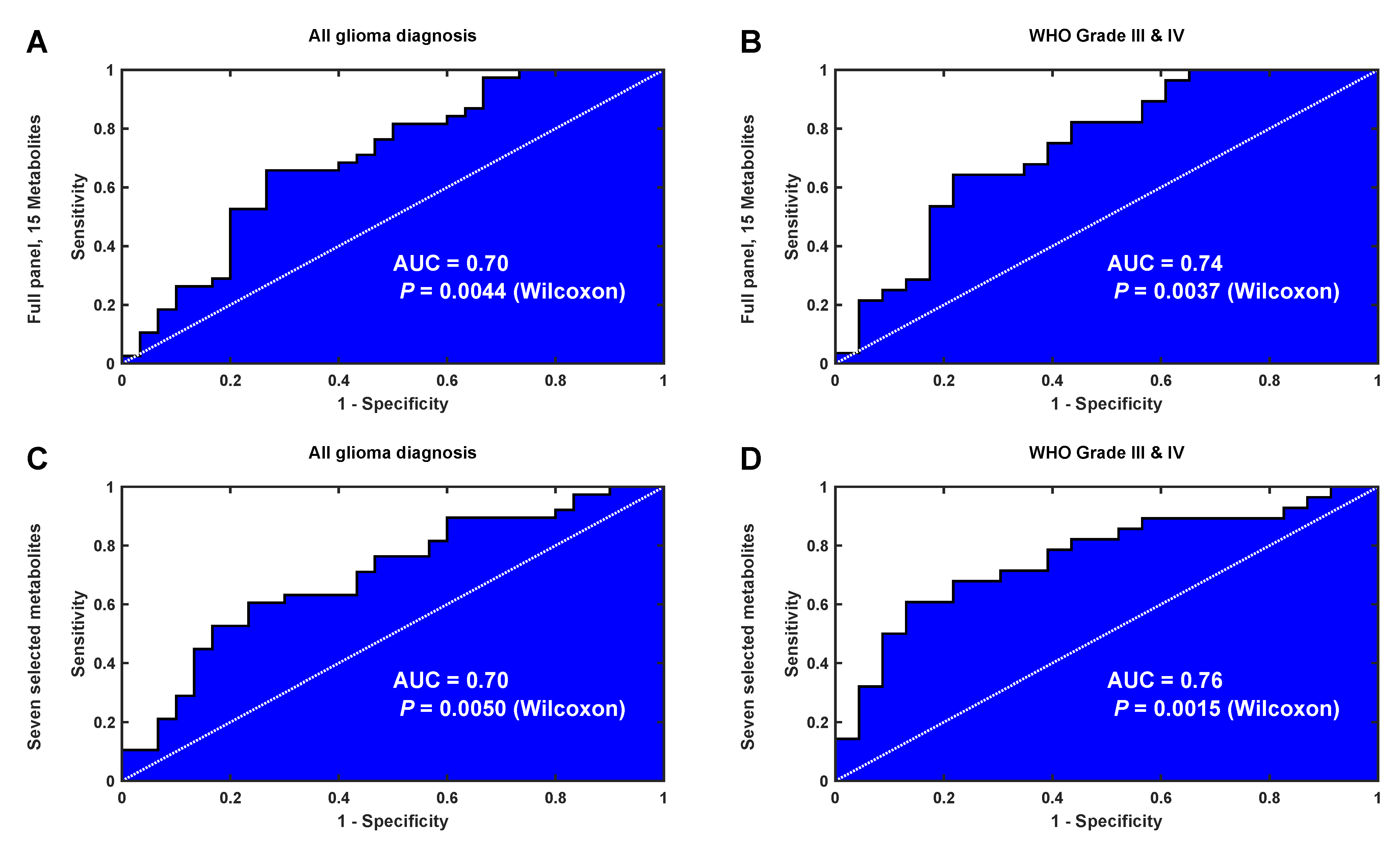
2.5. Validation of the Identified Latent Biomarker for Glioma
3. Discussion
4. Materials and Methods
4.1. Study Subjects and Sample Acquisition
4.2. Special Reagents
4.3. Metabolite Extraction and Analysis
4.4. Metabolite Identification and Quantification
4.5. Multivariate Statistical Modeling
4.6. Visualization of Molecular Networks
4.7. Data Accessibility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Westphal, M.; Lamszus, K. Circulating biomarkers for gliomas. Nat. Rev. Neurol. 2015, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.B. Biomarkers as drug development tools: Discovery, validation, qualification and use. Nat. Rev. Rheumatol. 2018, 14, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.; Wuolikainen, A.; Thysell, E.; Chorell, E.; Stattin, P.; Wikstrom, P.; Antti, H. Constrained randomization and multivariate effect projections improve information extraction and biomarker pattern discovery in metabolomics studies involving dependent samples. Metab. Off. J. Metab. Soc. 2015, 11, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemometr. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Körber, V.; Yang, J.; Barah, P.; Wu, Y.; Stichel, D.; Gu, Z.; Fletcher, M.N.C.; Jones, D.; Hentschel, B.; Lamszus, K.; et al. Evolutionary Trajectories of IDH(WT) Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell 2019, 35, 692–704. [Google Scholar] [CrossRef]
- Björkblom, B.; Wibom, C.; Jonsson, P.; Moren, L.; Andersson, U.; Johannesen, T.B.; Langseth, H.; Antti, H.; Melin, B. Metabolomic screening of pre-diagnostic serum samples identifies association between alpha- and gamma-tocopherols and glioblastoma risk. Oncotarget 2016, 7, 37043–37053. [Google Scholar] [CrossRef]
- Mören, L.; Bergenheim, A.T.; Ghasimi, S.; Brannstrom, T.; Johansson, M.; Antti, H. Metabolomic Screening of Tumor Tissue and Serum in Glioma Patients Reveals Diagnostic and Prognostic Information. Metabolites 2015, 5, 502–520. [Google Scholar] [CrossRef]
- Castillo, M.; Smith, J.K.; Kwock, L. Correlation of myo-inositol levels and grading of cerebral astrocytomas. AJNR. Am. J. Neuroradiol. 2000, 21, 1645–1649. [Google Scholar]
- Mören, L.; Wibom, C.; Bergstrom, P.; Johansson, M.; Antti, H.; Bergenheim, A.T. Characterization of the serum metabolome following radiation treatment in patients with high-grade gliomas. Radiat. Oncol. 2016, 11, 51. [Google Scholar] [CrossRef]
- Kallenberg, K.; Bock, H.C.; Helms, G.; Jung, K.; Wrede, A.; Buhk, J.H.; Giese, A.; Frahm, J.; Strik, H.; Dechent, P.; et al. Untreated glioblastoma multiforme: Increased myo-inositol and glutamine levels in the contralateral cerebral hemisphere at proton MR spectroscopy. Radiology 2009, 253, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Durmo, F.; Rydelius, A.; Baena, S.C.; Askaner, K.; Latt, J.; Bengzon, J.; Englund, E.; Chenevert, T.L.; Bjorkman-Burtscher, I.M.; Sundgren, P.C. Multivoxel (1)H-MR Spectroscopy Biometrics for Preoprerative Differentiation Between Brain Tumors. Tomography 2018, 4, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Björkblom, B.; Jonsson, P.; Tabatabaei, P.; Bergstrom, P.; Johansson, M.; Asklund, T.; Bergenheim, A.T.; Antti, H. Metabolic response patterns in brain microdialysis fluids and serum during interstitial cisplatin treatment of high-grade glioma. Br. J. Cancer 2020, 122, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D. Intracellular organic osmolytes: Function and regulation. J. Biol. Chem. 2008, 283, 7309–7313. [Google Scholar] [CrossRef]
- Haneda, M.; Kikkawa, R.; Arimura, T.; Ebata, K.; Togawa, M.; Maeda, S.; Sawada, T.; Horide, N.; Shigeta, Y. Glucose inhibits myo-inositol uptake and reduces myo-inositol content in cultured rat glomerular mesangial cells. Metab. Clin. Exp. 1990, 39, 40–45. [Google Scholar] [CrossRef]
- Olgemoller, B.; Schwaabe, S.; Schleicher, E.D.; Gerbitz, K.D. Upregulation of myo-inositol transport compensates for competitive inhibition by glucose. An explanation for the inositol paradox? Diabetes 1993, 42, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Yorek, M.A.; Dunlap, J.A.; Ginsberg, B.H. Effect of sorbinil on myo-inositol metabolism in cultured neuroblastoma cells exposed to increased glucose levels. J. Neurochem. 1988, 51, 331–338. [Google Scholar] [CrossRef]
- Andrisic, L.; Dudzik, D.; Barbas, C.; Milkovic, L.; Grune, T.; Zarkovic, N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018, 14, 47–58. [Google Scholar] [CrossRef]
- Panosyan, E.H.; Lin, H.J.; Koster, J.; Lasky, J.L., 3rd. In search of druggable targets for GBM amino acid metabolism. BMC Cancer 2017, 17, 162. [Google Scholar] [CrossRef]
- Carter, D.R.; Sutton, S.K.; Pajic, M.; Murray, J.; Sekyere, E.O.; Fletcher, J.; Beckers, A.; De Preter, K.; Speleman, F.; George, R.E.; et al. Glutathione biosynthesis is upregulated at the initiation of MYCN-driven neuroblastoma tumorigenesis. Mol. Oncol. 2016, 10, 866–878. [Google Scholar] [CrossRef]
- den Hartog, G.J.; Boots, A.W.; Adam-Perrot, A.; Brouns, F.; Verkooijen, I.W.; Weseler, A.R.; Haenen, G.R.; Bast, A. Erythritol is a sweet antioxidant. Nutrition 2010, 26, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.; Baynes, J.W.; Spiteller, G. The reaction of hyaluronic acid and its monomers, glucuronic acid and N-acetylglucosamine, with reactive oxygen species. Carbohydr. Res. 1999, 321, 228–234. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Degradation of hyaluronic acid, poly- and monosaccharides, and model compounds by hypochlorite: Evidence for radical intermediates and fragmentation. Free Radic. Biol. Med. 1998, 24, 1396–1410. [Google Scholar] [CrossRef]
- Soltes, L.; Kogan, G.; Stankovska, M.; Mendichi, R.; Rychly, J.; Schiller, J.; Gemeiner, P. Degradation of high-molar-mass hyaluronan and characterization of fragments. Biomacromolecules 2007, 8, 2697–2705. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Barrera, D.; Medina-Campos, O.N.; Carvajal, R.C.; Hernandez-Pando, R.; Macias-Ruvalcaba, N.A.; Maldonado, P.D.; Salcedo, M.I.; Tapia, E.; Saldivar, L.; et al. Time course study of oxidative and nitrosative stress and antioxidant enzymes in K2Cr2O7-induced nephrotoxicity. BMC Nephrol. 2005, 6, 4. [Google Scholar] [CrossRef]
- Ichida, K.; Amaya, Y.; Okamoto, K.; Nishino, T. Mutations associated with functional disorder of xanthine oxidoreductase and hereditary xanthinuria in humans. Int. J. Mol. Sci. 2012, 13, 15475–15495. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Petrotto, E.; Tozzi, M.G. Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. Int. J. Mol. Sci. 2018, 19, 3598. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase in cancer: More than a differentiation marker. Cancer Med. 2016, 5, 546–557. [Google Scholar] [CrossRef]
- Kokoglu, E.; Belce, A.; Ozyurt, E.; Tepeler, Z. Xanthine oxidase levels in human brain tumors. Cancer Lett. 1990, 50, 179–181. [Google Scholar] [CrossRef]
- Alvarez-Lario, B.; Macarron-Vicente, J. Is there anything good in uric acid? QJM Mon. J. Assoc. Physicians 2011, 104, 1015–1024. [Google Scholar] [CrossRef]
- Amirian, E.S.; Ostrom, Q.T.; Armstrong, G.N.; Lai, R.K.; Gu, X.; Jacobs, D.I.; Jalali, A.; Claus, E.B.; Barnholtz-Sloan, J.S.; Il’yasova, D.; et al. Aspirin, NSAIDs, and Glioma Risk: Original Data from the Glioma International Case-Control Study and a Meta-analysis. Cancer Epidemiol. Prev. Biomark. 2019, 28, 555–562. [Google Scholar] [CrossRef]
- Egan, K.M.; Nabors, L.B.; Thompson, Z.J.; Rozmeski, C.M.; Anic, G.A.; Olson, J.J.; LaRocca, R.V.; Chowdhary, S.A.; Forsyth, P.A.; Thompson, R.C. Analgesic use and the risk of primary adult brain tumor. Eur. J. Epidemiol. 2016, 31, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Yang, T.; Gan, Y.; Li, W.; Wang, C.; Gong, Y.; Lu, Z. Associations between aspirin use and the risk of cancers: A meta-analysis of observational studies. BMC Cancer 2018, 18, 288. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Wang, J.; Xie, L.; Li, T.; He, Y.; Peng, Q.; Qin, X.; Li, S. Association between nonsteroidal anti-inflammatory drug use and brain tumour risk: A meta-analysis. Br. J. Clin. Pharmacol. 2014, 78, 58–68. [Google Scholar] [CrossRef]
- Seliger, C.; Ricci, C.; Meier, C.R.; Bodmer, M.; Jick, S.S.; Bogdahn, U.; Hau, P.; Leitzmann, M.F. Diabetes, use of antidiabetic drugs, and the risk of glioma. Neuro-oncology 2016, 18, 340–349. [Google Scholar] [CrossRef]
- Amirian, E.S.; Zhou, R.; Wrensch, M.R.; Olson, S.H.; Scheurer, M.E.; Il’yasova, D.; Lachance, D.; Armstrong, G.N.; McCoy, L.S.; Lau, C.C.; et al. Approaching a Scientific Consensus on the Association between Allergies and Glioma Risk: A Report from the Glioma International Case-Control Study. Cancer Epidemiol. Prev. Biomark. 2016, 25, 282–290. [Google Scholar] [CrossRef]
- Schlehofer, B.; Blettner, M.; Preston-Martin, S.; Niehoff, D.; Wahrendorf, J.; Arslan, A.; Ahlbom, A.; Choi, W.N.; Giles, G.G.; Howe, G.R.; et al. Role of medical history in brain tumour development. Results from the international adult brain tumour study. Int. J. Cancer 1999, 82, 155–160. [Google Scholar] [CrossRef]
- Jonsson, P.; Johansson, E.S.; Wuolikainen, A.; Lindberg, J.; Schuppe-Koistinen, I.; Kusano, M.; Sjostrom, M.; Trygg, J.; Moritz, T.; Antti, H. Predictive metabolite profiling applying hierarchical multivariate curve resolution to GC-MS data—A potential tool for multi-parametric diagnosis. J. Proteome Res. 2006, 5, 1407–1414. [Google Scholar] [CrossRef]
- Eriksson, L.; Trygg, J.; Wold, S. CV-ANOVA for significance testing of PLS and OPLS (R) models. J. Chemometr. 2008, 22, 594–600. [Google Scholar] [CrossRef]
- Jonsson, P.; Björkblom, B.; Chorell, E.; Olsson, T.; Antti, H. Statistical loadings and latent significance simplify and improve interpretation of multivariate projection models. bioRxiv 2018. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wrzodek, C.; Buchel, F.; Ruff, M.; Drager, A.; Zell, A. Precise generation of systems biology models from KEGG pathways. BMC Syst. Biol. 2013, 7, 15. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Baseline Time Point Only (b | Repeated Time Point Only | Progression Pattern | |||
|---|---|---|---|---|---|---|
| Model | OPLS-DA | OPLS-EP | OPLS-DA | OPLS-EP | OPLS-DA | OPLS-EP |
| Observations | 128 | 64 | 128 | 64 | 128 | 64 |
| Components | - | - | 1 | 2 | 1 | 1 |
| R2Y | - | - | 0.12 | 0.3 | 0.12 | 0.34 |
| Q2 | - | - | −0.28 | 0.01 | −0.12 | 0.11 |
| P-value (CV-ANOVA) | - | - | 1 | 0.96 | 1 | 0.026 |
| Metabolite | HMDB ID | OPLS Loadings w (a | OPLS Loadings p (b | ||
|---|---|---|---|---|---|
| t-Value | P-Value | t-Value | P-Value | ||
| myo-Inositol | HMDB0000211 | 4.37 | 0.0002 | 7.73 | <0.0001 |
| scyllo-Inositol | HMDB0006088 | 2.08 | 0.047 | 3.08 | 0.0047 |
| Cysteine | HMDB0000574 | 2.61 | 0.014 | 2.94 | 0.0067 |
| Glycine | HMDB0000123 | 3.10 | 0.0044 | 2.83 | 0.0087 |
| Glyceric acid | HMDB0000139 | 2.35 | 0.026 | 2.61 | 0.015 |
| Aceturic acid (N-acetylglycine) | HMDB0000532 | 2.84 | 0.0083 | 2.36 | 0.026 |
| Phosphate (phosphoric acid) | HMDB0002142 | 2.62 | 0.014 | 4.26 | 0.0002 |
| Proline | HMDB0000162 | 2.43 | 0.022 | 4.11 | 0.0003 |
| 4-Hydroxyphenylacetic acid | HMDB0000020 | 2.25 | 0.032 | 3.31 | 0.0027 |
| Erythronic acid | HMDB0000613 | 3.11 | 0.0043 | 4.12 | 0.0003 |
| Erythritol | HMDB0002994 | 2.66 | 0.013 | 3.70 | 0.001 |
| N-acetylglucosamine (GlcNAc) | HMDB0000215 | 2.57 | 0.016 | 4.05 | 0.0004 |
| Creatinine | HMDB0000562 | 2.37 | 0.025 | 2.37 | 0.025 |
| Uric acid (urate) | HMDB0000289 | 2.09 | 0.046 | 3.20 | 0.0035 |
| Urea | HMDB0000294 | 2.17 | 0.039 | 3.12 | 0.0043 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonsson, P.; Antti, H.; Späth, F.; Melin, B.; Björkblom, B. Identification of Pre-Diagnostic Metabolic Patterns for Glioma Using Subset Analysis of Matched Repeated Time Points. Cancers 2020, 12, 3349. https://doi.org/10.3390/cancers12113349
Jonsson P, Antti H, Späth F, Melin B, Björkblom B. Identification of Pre-Diagnostic Metabolic Patterns for Glioma Using Subset Analysis of Matched Repeated Time Points. Cancers. 2020; 12(11):3349. https://doi.org/10.3390/cancers12113349
Chicago/Turabian StyleJonsson, Pär, Henrik Antti, Florentin Späth, Beatrice Melin, and Benny Björkblom. 2020. "Identification of Pre-Diagnostic Metabolic Patterns for Glioma Using Subset Analysis of Matched Repeated Time Points" Cancers 12, no. 11: 3349. https://doi.org/10.3390/cancers12113349
APA StyleJonsson, P., Antti, H., Späth, F., Melin, B., & Björkblom, B. (2020). Identification of Pre-Diagnostic Metabolic Patterns for Glioma Using Subset Analysis of Matched Repeated Time Points. Cancers, 12(11), 3349. https://doi.org/10.3390/cancers12113349