BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives



Abstract
Simple Summary
Abstract
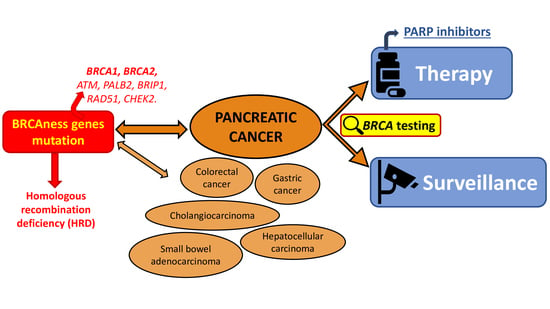
1. Introduction
2. Pancreatic Cancer
2.1. BRCA Testing for Therapeutic Purpose
2.2. BRCA Testing for Surveillance Purposes
3. Other Gastrointestinal Cancers
3.1. Colorectal Cancer
3.2. Gastric Cancer
3.3. Cholangiocarcinoma and Hepatocellular Carcinoma
3.4. Gastrointestinal Cancer Minorities
4. Future Perspectives
5. Conclusions
Funding
Conflicts of Interest
References
- Toss, A.; Cortesi, L. Molecular Mechanisms of PARP Inhibitors in BRCA-related Ovarian Cancer. J. Cancer Sci. Ther. 2013, 5, 409–416. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Stoppa-Lyonnet, D. The biological effects and clinical implications of BRCA mutations: Where do we go from here? Eur. J. Hum. Genet. 2016, 24, S3–S9. [Google Scholar] [CrossRef]
- Toss, A.; Molinaro, E.; Sammarini, M.; Del Savio, M.C.; Cortesi, L.; Facchinetti, F.; Grandi, G. Hereditary ovarian cancers: State of the art. Minerva Med. 2019, 110, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; De Matteis, E.; Toss, A.; Marchi, I.; Medici, V.; Contu, G.; Xholli, A.; Grandi, G.; Cagnacci, A.; Federico, M. Evaluation of Transvaginal Ultrasound plus CA-125 Measurement and Prophylactic Salpingo-Oophorectomy in Women at Different Risk Levels of Ovarian Cancer: The Modena Study Group Cohort Study. Oncology 2017, 93, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Tyrer, J.; Duffy, S.W.; Cuzick, J. A breast cancer prediction model incorporating familial and personal risk factors. Stat. Med. 2004, 23, 1111–1130. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.D.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nat. Cell Biol. 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association Between BRCA1 and BRCA2 Mutations and Survival in Women with Invasive Epithelial Ovarian Cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef]
- Kurnit, K.; Coleman, R.L.; Westin, S.N. Using PARP Inhibitors in the Treatment of Patients with Ovarian Cancer. Curr. Treat. Opt. Oncol. 2018, 19, 1. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Toss, A.; Venturelli, M.; Molinaro, E.; Pipitone, S.; Barbieri, E.; Marchi, I.; Tenedini, E.; Artuso, L.; Castellano, S.; Marino, M.; et al. Hereditary Pancreatic Cancer: A Retrospective Single-Center Study of 5143 Italian Families with History of BRCA-Related Malignancies. Cancers 2019, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, V.; Leslie, G.; Barnes, D.R.; Agnarsson, B.A.; Aittomäki, K.; Alducci, E.; Andrulis, I.L.; Barkardottir, R.B.; Barroso, A.; CIMBA Group; et al. Characterization of the Cancer Spectrum in Men with Germline BRCA1 and BRCA2 Pathogenic Variants: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). JAMA Oncol. 2020, 6, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Vrieling, A.; Lubin, J.H.; Kraft, P.; Mendelsohn, J.B.; Hartge, P.; Canzian, F.; Steplowski, E.; Arslan, A.A.; Gross, M.; et al. Cigarette Smoking and Pancreatic Cancer: A Pooled Analysis from the Pancreatic Cancer Cohort Consortium. Am. J. Epidemiol. 2009, 170, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Iodice, S.; Gandini, S.; Maisonneuve, P.; Lowenfels, A.B. Tobacco and the risk of pancreatic cancer: A review and meta-analysis. Langenbeck Arch. Surg. 2008, 393, 535–545. [Google Scholar] [CrossRef]
- Everhart, J.; Wright, D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA 1995, 273, 1605–1609. [Google Scholar] [CrossRef]
- Jiao, L.; Berrington de Gonzalez, A.; Hartge, P.; Pfeiffer, R.M.; Park, Y.; Freedman, D.M.; Gail, M.H.; Alavanja, M.C.; Albanes, D.; Beane Freeman, L.E.; et al. Body mass index, effect modifiers, and risk of pancreatic cancer: A pooled study of seven prospective cohorts. Cancer Causes Control. 2010, 21, 1305–1314. [Google Scholar] [CrossRef]
- Klein, A.P. Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 2012, 51, 14–24. [Google Scholar] [CrossRef]
- Welinsky, S.; Lucas, A.L. Familial Pancreatic Cancer and the Future of Directed Screening. Gut Liver 2017, 11, 761–770. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Takaori, K.; Morizane, C.; Maguchi, H.; Mizuma, M.; Takahashi, H.; Wada, K.; Hosoi, H.; Yachida, S.; Suzuki, M.; et al. Familial pancreatic cancer: Concept, management and issues. World J. Gastroenterol. 2017, 23, 935–948. [Google Scholar] [CrossRef]
- Ohmoto, A.; Yachida, S.; Morizane, C. Genomic Features and Clinical Management of Patients with Hereditary Pancreatic Cancer Syndromes and Familial Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 561. [Google Scholar] [CrossRef] [PubMed]
- Rebelatto, T.F.; Falavigna, M.; Pozzari, M.; Spada, F.; Cella, C.A.; Laffi, A.; Pellicori, S.; Fazio, N. Should platinum-based chemotherapy be preferred for germline BReast CAncer genes (BRCA) 1 and 2-mutated pancreatic ductal adenocarcinoma (PDAC) patients? A systematic review and meta-analysis. Cancer Treat. Rev. 2019, 80, 101895. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.L.; Tseng, J.F.; Miksad, R.A. Genetic Testing in Pancreatic Ductal Adenocarcinoma: Implications for Prevention and Treatment. Clin Ther. 2016, 38, 1622–1635. [Google Scholar] [CrossRef]
- Young, E.L.; Thompson, B.A.; Neklason, D.W.; Firpo, M.A.; Werner, T.; Bell, R.; Berger, J.; Fraser, A.; Gammon, A.; Koptiuch, C.; et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer 2018, 18, 697. [Google Scholar] [CrossRef]
- Iqbal, J.; Ragone, A.V.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.Z.; Neuhausen, S.L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- The Breast Cancer Linkage Consortium. Cancer Risks in BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.E.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2015, 6, 166–175. [Google Scholar] [CrossRef]
- Reiss, K.A.; Yu, S.; Judy, R.; Symecko, H.; Nathanson, K.L.; Domchek, S.M. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef]
- Golan, T.; Sella, T.; O’Reilly, E.; Katz, M.H.G.; Epelbaum, R.; Kelsen, D.P.; Borgida, A.; Maynard, H.; Kindler, H.; Friedmen, E.; et al. Overall survival and clinical characteristics of BRCA mutation carriers with stage I/II pancreatic cancer. Br. J. Cancer 2017, 116, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Groot, V.P.; Gemenetzis, G.; Wei, J.; Cameron, J.L.; Weiss, M.J.; Goggins, M.; Wolfgang, C.L.; Yu, J.; He, J. BRCA1/BRCA2 Germline Mutation Carriers and Sporadic Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2018, 226, 630–637.e1. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Initiative, A.P.C.G.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nat. Cell Biol. 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Pant, S.; Maitra, A.; Yap, T.A. PARP inhibition—Opportunities in pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 16, 595–596. [Google Scholar] [CrossRef]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase Inhibitors in Pancreatic Cancer: A New Treatment Paradigms and Future Implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wilkerson, P.M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J.S. Synthetic lethality of PARP inhibition in cancers lackingBRCA1andBRCA2mutations. Cell Cycle 2011, 10, 1192–1199. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Hammel, P.; Kindler, H.L.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Health-related quality of life in patients with a germline BRCA mutation and metastatic pancreatic cancer receiving maintenance olaparib. Ann. Oncol. 2019, 30, 1959–1968. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2019, 122, 333–339. [Google Scholar] [CrossRef]
- Lowery, M.A. Genotype–phenotype correlation in BRCA1/2 mutation-associated pancreatic cancer. Br. J. Cancer 2019, 122, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.M.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Hongkun Wang, H.; Parenti, S.; He, A.R.; Hwang, J.J.; Ley, L.; Difebo, H.; Smaglo, B.G.; Kim, S.S.; Weinberg, B.A.; et al. Final report of a phase I/II study of veliparib (Vel) in combination with 5-FU and oxaliplatin (FOLFOX) in patients (pts) with metastatic pancreatic cancer (mPDAC). J. Clin. Oncol. 2019, 37, 4015. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Guthrie, K.A.; Philip, P.A.; Swisher, E.M.; Jalikis, F.; Pishvaian, M.J.; Berlin, J.; Noel, M.S.; Suga, J.M.; Garrido-Laguna, I.; et al. Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. J. Clin. Oncol. 2019, 37, 4014. [Google Scholar] [CrossRef]
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-Time Genomic Profiling of Pancreatic Ductal Adenocarcinoma: Potential Actionability and Correlation with Clinical Phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef]
- Wong, W.; Raufi, A.G.; Safyan, R.A.; Bates, S.E.; Manji, G.A. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Manag. Res. 2020, 12, 2731–2742. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Chiorean, E.G.; Czito, B.; Scaife, C.; Narang, A.K.; Fountzilas, C.; Wolpin, B.M.; Al-Hawary, M.; Asbun, H.; et al. Guidelines Insights: Pancreatic Adenocarcinoma, Version 1.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 202–210. [Google Scholar] [CrossRef]
- Stoffel, E.M.; McKernin, S.E.; Brand, R.; Canto, M.; Goggins, M.; Moravek, C.; Nagarajan, A.; Petersen, G.M.; Simeone, D.M.; Yurgelun, M.; et al. Evaluating Susceptibility to Pancreatic Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2019, 37, 153–164. [Google Scholar] [CrossRef]
- Vasen, H.F.; Ibrahim, I.; Ponce, C.G.; Slater, E.P.; Matthäi, E.; Carrato, A.; Earl, J.; Robbers, K.; Van Mil, A.M.; Potjer, T.; et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies from Three European Expert Centers. J. Clin. Oncol. 2016, 34, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Paiella, S.; Capurso, G.; Cavestro, G.M.; Butturini, G.; Pezzilli, R.; Salvia, R.; Signoretti, M.; Crippa, S.; Carrara, S.; Frigerio, I.; et al. Results of First-Round of Surveillance in Individuals at High-Risk of Pancreatic Cancer from the AISP (Italian Association for the Study of the Pancreas) Registry. Am. J. Gastroenterol. 2019, 114, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Paiella, S.; Salvia, R.; De Pastena, M.; Pollini, T.; Casetti, L.; Landoni, L.; Esposito, A.; Marchegiani, G.; Malleo, G.; De Marchi, G.; et al. Screening/surveillance programs for pancreatic cancer in familial high-risk individuals: A systematic review and proportion meta-analysis of screening results. Pancreatology 2018, 18, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Signoretti, M.; Bruno, M.J.; Zerboni, G.; Poley, J.-W.; Fave, G.D.; Capurso, G. Results of surveillance in individuals at high-risk of pancreatic cancer: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2018, 6, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Almario, J.A.; Schulick, R.D.; Yeo, C.J.; Klein, A.; Blackford, A.; Shin, E.J.; Sanyal, A.; Yenokyan, G.; Lennon, A.M.; et al. Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-term Surveillance. Gastroenterology 2018, 155, 740–751.e2. [Google Scholar] [CrossRef] [PubMed]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2019, 69, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, N.; Moreira, L.; Carneiro, F.; Balaguer, F.; Cervantes, A.; Balmaña, J.; Martinelli, E. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1558–1571. [Google Scholar] [CrossRef]
- Lu, C.; Xu, C.F.; Wan, X.Y.; Zhu, H.T.; Yu, C.H.; Li, Y.M. Screening for pancreatic cancer in familial high-risk individuals: A systematic review. World J. Gastroenterol. 2015, 21, 8678–8686. [Google Scholar] [CrossRef]
- Konings, I.C.A.W.; Sidharta, G.N.; Harinck, F.; Aalfs, C.M.; Poley, J.W.; Kieffer, J.M.; Kuenen, M.A.; Smets, E.M.A.; Wagner, A.; Van Hooft, J.E.; et al. Repeated participation in pancreatic cancer surveillance by high-risk individuals imposes low psychological burden. Psycho Oncol. 2015, 25, 971–978. [Google Scholar] [CrossRef]
- Paiella, S.; Marinelli, V.; Secchettin, E.; Mazzi, M.A.; Ferretto, F.; Casolino, R.; Bassi, C.; Salvia, R. The emotional impact of surveillance programs for pancreatic cancer on high-risk individuals: A prospective analysis. Psycho Oncol. 2020, 29, 1004–1011. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, M.W.; Magnuson, C.W.; Larsen, A.L.; Krush, A.J. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch. Intern. Med. 1966, 117, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Karstensen, J.G.; Burisch, J.; Pommergaard, H.-C.; Aalling, L.; Højen, H.; Jespersen, N.; Schmidt, P.N.; Bülow, S. Colorectal Cancer in Individuals with Familial Adenomatous Polyposis, Based on Analysis of the Danish Polyposis Registry. Clin. Gastroenterol. Hepatol. 2019, 17, 2294–2300.e1. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.F.; Bishop, D.T.; Narod, S.A.; Goldgar, D.E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994, 343, 692–695. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.F. Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer Risk Estimates for BRCA1 Mutation Carriers Identified in a Risk Evaluation Program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2013, 110, 530–534. [Google Scholar] [CrossRef]
- Suchy, J.; Cybulski, C.; Górski, B.; Huzarski, T.; Byrski, T.; Dębniak, T.; Gronwald, J.; Jakubowska, A.; Wokołorczyk, M.; Kurzawski, G.; et al. BRCA1 mutations and colorectal cancer in Poland. Fam. Cancer 2010, 9, 541–544. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.-E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Burn, J.; Gerdes, A.-M.; Macrae, F.A.; Mecklin, J.-P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- US Preventive Services Task Force. Routine aspirin or nonsteroidal anti-inflammatory drugs for the primary prevention of colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2007, 146, 361–364. [Google Scholar] [CrossRef]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated withBRCA1andBRCA2mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.M.; Ternent, C.A.; Adams, D.R.; Thorson, A.G.; Blatchford, G.J.; Christensen, M.A.; Watson, P.; Lynch, H.T. Colorectal cancer in hereditary breast cancer kindreds. Dis. Colon Rectum 1999, 42, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.M.; Ternent, C.A.; Adams, D.R.; Thorson, A.G.; Blatchford, G.J.; Christensen, M.A.; Watson, P.; Lynch, H.T. BRCA1 and BRCA2 founder mutations and the risk of colorectal cancer. J. Natl Cancer Inst. 2004, 96, 15–21. [Google Scholar]
- Slattery, M.L.; Kerber, R.A. Family History of Cancer and Colon Cancer Risk: The Utah Population Database. J. Natl. Cancer Inst. 1994, 86, 1618–1626. [Google Scholar] [CrossRef]
- Woodage, T.; King, S.M.; Wacholder, S.; Hartge, P.; Struewing, J.P.; McAdams, M.; Laken, S.J.; Tucker, M.A.; Brody, L.C. The APCI1307K allele and cancer risk in a community-based study of Ashkenazi Jews. Nat. Genet. 1998, 20, 62–65. [Google Scholar] [CrossRef]
- Redston, M.; Nathanson, K.L.; Yuan, Z.Q.; Neuhausen, S.L.; Satagopan, J.M.; Wong, N.; Yang, D.; Nafa, D.; Abrahamson, J.; Ozcelik, H.; et al. The APC I1307K allele and breast cancer risk. Nat. Genet. 1998, 20, 13–14. [Google Scholar] [CrossRef]
- Oh, M.; McBride, A.; Yun, S.; Bhattacharjee, S.; Slack, M.; Jeter, J.M.; Abraham, I. BRCA1 and BRCA2 gene mutations and colorectal cancer risk: Systematic review and meta-analysis. J. Clin. Oncol. 2018, 36, 605. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Peng, L.; Zhang, D.; Jin, M.; Wang, J.; Xue, J.; Liu, H.; Zhang, T. Complete pathological response following neoadjuvant FOLFOX chemotherapy in BRCA2-mutant locally advanced rectal cancer: A case report. BMC Cancer 2018, 18, 1253. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.-M.B.; Fox, S.; et al. Combined immune checkpoint blockade as a therapeutic strategy forBRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Naseem, M.; Xiu, J.; Salem, M.E.; Goldberg, R.M.; VanderWalde, A.M.; Grothey, A.; Philip, P.A.; Seeber, A.; Puccini, A.; Tokunaga, R.; et al. Characteristics of colorectal cancer (CRC) patients with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2019, 37, 606. [Google Scholar] [CrossRef]
- Harpaz, N.; Gatt, Y.E.; Granit, R.Z.; Fruchtman, H.; Hubert, A.; Grinshpun, A. Mucinous Histology, BRCA1/2 Mutations, and Elevated Tumor Mutational Burden in Colorectal Cancer. J. Oncol. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Davidson, D.; Wang, Y.; Aloyz, R.; Panasci, L. The PARP inhibitor ABT-888 synergizes irinotecan treatment of colon cancer cell lines. Investig. New Drugs 2012, 31, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.W.; Waxweiler, T.V.; Landry, J.; Gao, H.; Xu, Y.; Wang, L.; El-Rayes, B.; Shu, H.-K.G. In Vitro and In Vivo Enhancement of Chemoradiation Using the Oral PARP Inhibitor ABT-888 in Colorectal Cancer Cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 469–476. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mohamedali, A.M.; Patterson, M.; Matto, N.; Smith, A.; Kulasekararaj, A.; Chelliah, R.; Curtin, N.J.; Farzaneh, F.; Shall, S.; et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica 2013, 98, 1397–1406. [Google Scholar] [CrossRef]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.; et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncology 2016, 21, 172–177. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef]
- Tulinius, H.; Olafsdottir, G.H.; Sigvaldason, H.; Arason, A.; Barkardottir, R.B.; Egilsson, V.; Ogmundsdottir, H.M.; Tryggvadottir, L.; Gudlaugsdottir, S.; Eyfjord, J.E. The effect of a single BRCA2 mutation on cancer in Iceland. J. Med. Genet. 2002, 39, 457–462. [Google Scholar] [CrossRef]
- Van Asperen, C.J.; Brohet, R.M.; Meijers-Heijboer, E.J.; Hoogerbrugge, N.; Verhoef, S.; Vasen, H.F.A.; Ausems, M.G.E.M.; Menko, F.H.; Garcia, E.B.G.; Klijn, J.G.M.; et al. Cancer risks in BRCA2 families: Estimates for sites other than breast and ovary. J. Med. Genet. 2005, 42, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, J.L.; García-Pérez, A.; Hemminki, K. Contribution of the Defective BRCA1, BRCA2 and CHEK2 Genes to the Familial Aggregation of Breast Cancer: A Simulation Study Based on the Swedish Family-Cancer Database. Hered. Cancer Clin. Pr. 2004, 2, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- ClinicalTrials.gov Identifier NCT02734004. A Phase I/II Study of MEDI4736 in Combination with Olaparib in Patients with Advanced Solid Tumors. (MEDIOLA). Available online: https://clinicaltrials.gov/ct2/show/NCT02734004 (accessed on 2 September 2020).
- ClinicalTrials.gov Identifier NCT03008278. Olaparib and Ramucirumab in Treating Patients with Metastatic or Locally Recurrent Gastric or Gastroesophageal Junction Cancer That Cannot be Removed by Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT03008278 (accessed on 2 September 2020).
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncology 2017, 22, 804–810. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, J.; Qin, S.K.; Hua, H.Q. Treatment with olaparib monotherapy for BRCA2-mutated refractory intrahepatic cholangiocarcinoma: A case report. Onco Targets Ther. 2018, 11, 5957–5962. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.M.; Salem, M.E.; Battaglin, F.; et al. Frequency of BRCA mutation in biliary tract cancer and its correlation with tumor mutational burden (TMB) and microsatellite instability (MSI). J. Clin. Oncol. 2019, 37, 4085. [Google Scholar] [CrossRef]
- Fehling, S.C.; Miller, A.L.; Garcia, P.L.; Vance, R.B.; Yoon, K.J. The combination of BET and PARP inhibitors is synergistic in models of cholangiocarcinoma. Cancer Lett. 2020, 468, 48–58. [Google Scholar] [CrossRef]
- Mao, X.; Du, S.; Yang, Z.; Zhang, L.; Peng, X.; Jiang, N.; Zhou, H. Inhibitors of PARP-1 exert inhibitory effects on the biological characteristics of hepatocellular carcinoma cells in vitro. Mol. Med. Rep. 2017, 16, 208–214. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov Identifier NCT04042831. Olaparib in Treating Patients with Metastatic Biliary Tract Cancer with Aberrant DNA Repair Gene Mutations. Available online: https://clinicaltrials.gov/ct2/show/NCT04042831 (accessed on 2 September 2020).
- Lin, J.; Shi, J.; Guo, H.; Yang, X.; Jiang, Y.; Long, J.; Bai, Y.; Wang, D.; Yang, X.; Wan, X.; et al. Alterations in DNA Damage Repair Genes in Primary Liver Cancer. Clin. Cancer Res. 2019, 25, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, U.A.; Katainen, R.; Tanskanen, T.; Plaketti, R.-M.; Laine, R.; Hamberg, J.; Ristimäki, A.; Pukkala, E.; Taipale, M.; Mecklin, J.-P.; et al. Exome-wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet. 2018, 14, e1007200. [Google Scholar] [CrossRef] [PubMed]
- Quaas, A.; Heydt, C.; Waldschmidt, D.; Alakus, H.; Zander, T.; Goeser, T.; Kasper, P.; Bruns, C.J.; Brunn, A.; Roth, W.; et al. Alterations in ERBB2 and BRCA and microsatellite instability as new personalized treatment options in small bowel carcinoma. BMC Gastroenterol. 2019, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Beger, C.; Ramadani, M.; Meyer, S.; Leder, G.; Krüger, M.; Welte, K.; Gansauge, F.; Beger, H.G. Down-Regulation of BRCA1 in Chronic Pancreatitis and Sporadic Pancreatic Adenocarcinoma. Clin. Cancer Res. 2004, 10, 3780–3787. [Google Scholar] [CrossRef][Green Version]
- Golan, T.D.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluchshimon, S.; Kaufman, B.A.; Gershonibaruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Xu, R.-H.; Yu, X.; Hao, J.; Wang, L.; Pan, H.; Han, G.; Xu, J.; Zhang, Y.; Yang, S.; Chen, J.; et al. Efficacy and safety of weekly nab-paclitaxel plus gemcitabine in Chinese patients with metastatic adenocarcinoma of the pancreas: A phase II study. BMC Cancer 2017, 17, 885. [Google Scholar] [CrossRef]
- Kobayashi, N.; Shimamura, T.; Tokuhisa, M.; Goto, A.; Endo, I.; Ichikawa, Y. Effect of FOLFIRINOX as second-line chemotherapy for metastatic pancreatic cancer after gemcitabine-based chemotherapy failure. Medicine 2017, 96, e6769. [Google Scholar] [CrossRef]
- Soyano, A.E.; Baldeo, C.; Kasi, P.M. BRCA Mutation and Its Association with Colorectal Cancer. Clin. Color. Cancer 2018, 17, e647–e650. [Google Scholar] [CrossRef]
- Cercek, A.; Roxburgh, C.S.; Strombom, P.; Smith, J.J.; Temple, L.K.; Nash, G.M.; Guillem, J.G.; Paty, P.B.; Yaeger, R.; Stadler, Z.K.; et al. Adoption of Total Neoadjuvant Therapy for Locally Advanced Rectal Cancer. JAMA Oncol. 2018, 4, e180071. [Google Scholar] [CrossRef]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.-L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A Novel Bioavailable Inhibitor of Poly(ADP-ribose) Polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAPS Consortium Consensus for Surveillance of HRIs. |
|---|
| Individuals with at least three or more affected relatives, of whom at least one is an FDR to the individual considered for screening |
| Individuals with at least two affected relatives who are FDRs to each other, of whom at least one is an FDR to the individual considered for surveillance |
| Individuals with at least two affected relatives on the same side of the family, of whom at least one is an FDR to the individual considered for surveillance |
| LKB1/STK11 mutation carriers (PJS) regardless of family history |
| CDKN2A mutation carriers regardless of family history |
| BRCA1 mutation carriers with one affected FDR |
| BRCA2 mutation carriers with one affected FDR (or two affected family members, no FDR) with PC |
| PALB2 mutation carriers with one affected FDR |
| MMR gene mutation carriers (LS) with one affected FDR |
| ATM mutation carriers with one affected FDR |
| NCI Trial Number | Intervention | Cancer | Primary Endpoint | Phase |
|---|---|---|---|---|
| NCT03337087 | Nal-IRI+ Fluorouracil + Rucaparib | Pancreatic, colorectal, gastroesophageal or biliary cancer | Toxicity, ORR, best response rate | I, II |
| NCT03838406 | FOLFOX/CAPOX | Gastric cancer | ORR | Not Applicable |
| NCT03565991 | Talazoparib + Avelumab | Advanced solid tumors | OR | II |
| NCT02286687 | Talazoparib | Recurrent, refractory, advanced or metastatic cancers | Clinical benefit | II |
| NCT01989546 | BMN 673 (Talazoparib) | Advanced solid tumors | Pharmacodynamic effect of talazoparib; response rate | I, II |
| NCT03140670 | Rucaparib | Pancreatic cancer | Safety | II |
| NCT03428802 | Pembrolizumab | Advanced solid tumors | Response rate | II |
| NCT02723864 | Veliparib + VX-970 + Cisplatin | Refractory solid tumors | Safety, MTD, tolerability | I |
| NCT04182516 | NMS-03305293 | Advanced/metastatic solid tumors | Toxicity | I |
| NCT03875313 | CB-839 + Talazoparib | Solid tumors | Safety, MTD, tolerability | I, II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinaro, E.; Andrikou, K.; Casadei-Gardini, A.; Rovesti, G. BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives. Cancers 2020, 12, 3346. https://doi.org/10.3390/cancers12113346
Molinaro E, Andrikou K, Casadei-Gardini A, Rovesti G. BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives. Cancers. 2020; 12(11):3346. https://doi.org/10.3390/cancers12113346
Chicago/Turabian StyleMolinaro, Eleonora, Kalliopi Andrikou, Andrea Casadei-Gardini, and Giulia Rovesti. 2020. "BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives" Cancers 12, no. 11: 3346. https://doi.org/10.3390/cancers12113346
APA StyleMolinaro, E., Andrikou, K., Casadei-Gardini, A., & Rovesti, G. (2020). BRCA in Gastrointestinal Cancers: Current Treatments and Future Perspectives. Cancers, 12(11), 3346. https://doi.org/10.3390/cancers12113346