Breast Cancer Heterogeneity and Response to Novel Therapeutics


Simple Summary
Abstract
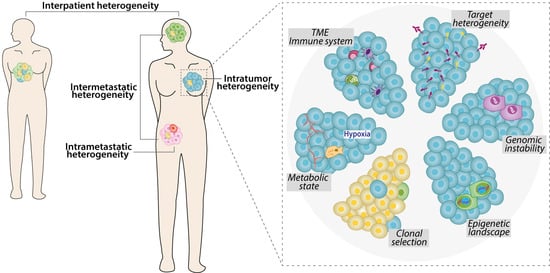
1. Phenotypic Features of Tumor Heterogeneity
2. Genomic Indications of Tumor Heterogeneity
3. Genetic Heterogeneity and Personalized Medicine
4. Heterogeneity and Resistance to Anti-Cancer Therapies
4.1. Heterogeneity in Target Expression
4.2. Clonal Selection and Resistance
4.3. Presence of Compensatory Signaling Pathways
4.4. Genomic Instability
4.5. Epigenetic Modifications
4.6. Tumor Microenvironment and Immune System
4.7. Metabolism
5. Beyond Tumor Heterogeneity
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Fidler, I.; Hart, I. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. Generation of phenotypic diversity and progression in metastatic tumor cells. Cancer Metastasis Rev. 1984, 3, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Cajal, S.R.Y.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Nassar, A.; Radhakrishnan, A.; Cabrero, I.A.; Cotsonis, G.A.; Cohen, C. Intratumoral Heterogeneity of Immunohistochemical Marker Expression in Breast Carcinoma: A tissue microarray-based study. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Pertschuk, L.P.; Axiotis, C.A.; Feldman, J.G.; Kim, Y.-D.; Karavattayhayyil, S.J.; Braithwaite, L. Marked Intratumoral Heterogeneity of the Proto-Oncogene Her-2/neu Determined by Three Different Detection Systems. Breast J. 1999, 5, 369–374. [Google Scholar] [CrossRef]
- Davis, B.W.; Zava, D.T.; Locher, G.W.; Goldhirsch, A.; Hartmann, W.H. Receptor heterogeneity of human breast cancer as measured by multiple intratumoral assays of estrogen and progesterone receptor. Eur. J. Cancer Clin. Oncol. 1984, 20, 375–382. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Gao, R.; Kim, C.; Sei, E.; Foukakis, T.; Crosetto, N.; Chan, L.-K.; Srinivasan, M.; Zhang, H.; Meric-Bernstam, F.; Navin, N.E. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 2017, 8, 228. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.; Rueda, O.M.; Vollan, H.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.; et al. The somatic mutation profiles of 2433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Baslan, T.; Kendall, J.; Volyanskyy, K.; McNamara, K.; Cox, H.; D’Italia, S.; Ambrosio, F.; Riggs, M.; Rodgers, L.; Leotta, A.; et al. Novel insights into breast cancer copy number genetic heterogeneity revealed by single-cell genome sequencing. eLife 2020, 9, e51480. [Google Scholar] [CrossRef]
- Condorelli, R.; Mosele, F.; Verret, B.; Bachelot, T.; Bedard, P.L.; Cortes, J.; Hyman, D.M.; Juric, D.; Krop, I.; Bieche, I.; et al. Genomic alterations in breast cancer: Level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2019, 30, 365–373. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The Genomic Landscapes of Human Breast and Colorectal Cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Kratz, J.; Burkard, M.; O’Meara, T.; Pusztai, L.; Veitch, Z.; Bedard, P.L. Incorporating Genomics Into the Care of Patients With Advanced Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Spira, A.; Yurgelun, M.B.; Alexandrov, L.; Rao, A.; Bejar, R.; Polyak, K.; Giannakis, M.; Shilatifard, A.; Finn, O.J.; Dhodapkar, M.; et al. Precancer Atlas to Drive Precision Prevention Trials. Cancer Res. 2017, 77, 1510–1541. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody–Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Trastuzumab Emtansine: Mechanisms of Action and Resistance, Clinical Progress, and Beyond. Trends Cancer 2020, 6, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocaña, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Cajal, S.R.Y.; Arribas, J.; et al. Expression of p95HER2, a Truncated Form of the HER2 Receptor, and Response to Anti-HER2 Therapies in Breast Cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Tsurutani, J.; Takahashi, S.; Iwata, H.; Krop, I.E.; Redfern, C.; Sagara, Y.; Doi, T.; Park, H.; Murthy, R.K.; et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: A dose-expansion, phase 1 study. Lancet Oncol. 2019, 20, 816–826. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Wang, T.; Wang, D.; Liu, J.; Feng, B.; Zhou, F.; Zhang, H.; Zhou, L.; Yin, Q.; Zhang, Z.; Cao, Z.; et al. Acidity-Triggered Ligand-Presenting Nanoparticles to Overcome Sequential Drug Delivery Barriers to Tumors. Nano Lett. 2017, 17, 5429–5436. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, H.; Brown, J.R.; Schalper, K.; Haack, H.; Silver, M.R.; Nixon, C.; Bossuyt, V.; Pusztai, L.; Lannin, D.R.; Rimm, D.L. PD-L1 Expression Correlates with Tumor-Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy in Breast Cancer. Cancer Immunol. Res. 2015, 3, 326–332. [Google Scholar] [CrossRef]
- Dill, E.A.; Gru, A.A.; Atkins, K.A.; Friedman, L.A.; Moore, M.E.; Bullock, T.N.; Cross, J.V.; Dillon, P.M.; Mills, A.M. PD-L1 Expression and Intratumoral Heterogeneity Across Breast Cancer Subtypes and Stages. Am. J. Surg. Pathol. 2017, 41, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. KEYNOTE-355: Randomized, double-blind, phase III study of pembrolizumab + chemotherapy versus placebo + chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer. J. Clin. Oncol. 2020, 38, 1000. [Google Scholar] [CrossRef]
- Pelekanou, V.; Barlow, W.E.; Nahleh, Z.A.; Wasserman, B.; Lo, Y.-C.; Von Wahlde, M.-K.; Hayes, D.; Hortobagyi, G.N.; Gralow, J.; Tripathy, D.; et al. Tumor-Infiltrating Lymphocytes and PD-L1 Expression in Pre- and Posttreatment Breast Cancers in the SWOG S0800 Phase II Neoadjuvant Chemotherapy Trial. Mol. Cancer Ther. 2018, 17, 1324–1331. [Google Scholar] [CrossRef]
- Li, X.; Warren, S.; Pelekanou, V.; Wali, V.; Cesano, A.; Liu, M.; Danaher, P.; Elliott, N.; Nahleh, Z.A.; Hayes, D.F.; et al. Immune profiling of pre- and post-treatment breast cancer tissues from the SWOG S0800 neoadjuvant trial. J. Immunother. Cancer 2019, 7, 88. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Hochhaus, A.; Erben, P.; Ernst, T.; Mueller, M.C. Resistance to Targeted Therapy in Chronic Myelogenous Leukemia. Semin. Hematol. 2007, 44, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Intermesoli, T.; Stefanoni, P.; Rambaldi, A. Mechanisms of Resistance to Targeted Therapies in Chronic Myeloid Leukemia. In Mechanisms of Drug Resistance in Cancer Therapy; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2017; Volume 249, pp. 231–250. [Google Scholar]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Brady, S.W.; McQuerry, J.A.; Qiao, Y.; Piccolo, S.R.; Shrestha, G.; Jenkins, D.F.; Layer, R.M.; Pedersen, B.S.; Miller, R.H.; Esch, A.; et al. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 2017, 8, 1231. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Chan, T.E.; Lombardo, Y.; Corleone, G.; Rotmensz, N.; Bravaccini, S.; Rocca, A.; Pruneri, G.; McEwen, K.R.; Coombes, R.C.; et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun. 2019, 10, 3840. [Google Scholar] [CrossRef]
- Burrell, R.A.; Swanton, C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C.; et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and Acquired Resistance to HER2-Targeted Therapies in HER2 Gene-Amplified Breast Cancer: Mechanisms and Clinical Implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Gandullo-Sánchez, L.; Capone, E.; Ocaña, A.; Iacobelli, S.; Sala, G.; Pandiella, A. HER3 targeting with an antibody-drug conjugate bypasses resistance to anti-HER2 therapies. EMBO Mol. Med. 2020, 12, e11498. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C. Signatures of DNA-Repair Deficiencies in Breast Cancer. N. Engl. J. Med. 2017, 377, 2490–2492. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ’BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Targeting the DNA repair defect of BRCA tumours. Curr. Opin. Pharmacol. 2005, 5, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP Inhibitors in Cancer Biology and Therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis-Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Risom, T.; Langer, E.M.; Chapman, M.P.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018, 9, 3815. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e7. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Jimenez, C.; Galan-Moya, E.M.; Corrales-Sanchez, V.; Noblejas-López, M.D.M.; Burgos, M.; Domingo, B.; Montero, J.C.; Gomez-Juarez, M.; Picazo-Martinez, M.G.; Esparis-Ogando, A.; et al. Inhibition of the mitotic kinase PLK1 overcomes therapeutic resistance to BET inhibitors in triple negative breast cancer. Cancer Lett. 2020, 491, 50–59. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. 2018, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Atabakhsh, E.; Kim, J.; Biette, K.M.; Van Rechem, C.; Ladd, B.; Burrowes, P.D.; Donado, C.; Mattoo, H.; Kleinstiver, B.P.; et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015, 29, 1018–1031. [Google Scholar] [CrossRef]
- Widmer, D.S.; Hoek, K.S.; Cheng, P.F.; Eichhoff, O.M.; Biedermann, T.; Raaijmakers, M.I.G.; Hemmi, S.; Dummer, R.; Levesque, M.P. Hypoxia Contributes to Melanoma Heterogeneity by Triggering HIF1α-Dependent Phenotype Switching. J. Investig. Dermatol. 2013, 133, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral Hypoxic Gradient Drives Stem Cells Distribution and MGMT Expression in Glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; DeMaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers From Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef]
- Ali, H.R.; Provenzano, E.; Dawson, S.-J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12 439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-Associated Lymphocytes As an Independent Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.-R.; Kurian, A.; Van Valen, D.; West, R.; et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018, 174, 1373–1387.e19. [Google Scholar] [CrossRef]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Rosario, A.; Wang, R.; Solomon, S.; Srinivasan, G.; Nelson, M.S.; Huang, Y.; Lim, M.H.; et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019, 4, e130000. [Google Scholar] [CrossRef]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.C.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2016, 47, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Zheng, P. Tumor cells versus host immune cells: Whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. 2018, 8, 34. [Google Scholar] [CrossRef]
- Noblejas-López, M.D.M.; Nieto-Jiménez, C.; García, S.M.; Pérez-Peña, J.; Nuncia-Cantarero, M.; Andrés-Pretel, F.; Galán-Moya, E.M.; Amir, E.; Pandiella, A.; Győrffy, B.; et al. Expression of MHC class I, HLA-A and HLA-B identifies immune-activated breast tumors with favorable outcome. OncoImmunology 2019, 8, e1629780. [Google Scholar] [CrossRef]
- Pérez-Pena, J.; Fekete, J.T.; Páez, R.; Baliu-Piqué, M.; García-Saenz, J.Á.; García-Barberán, V.; Manzano, A.; Pérez-Segura, P.; Esparis-Ogando, A.; Pandiella, A.; et al. A Transcriptomic Immunologic Signature Predicts Favorable Outcome in Neoadjuvant Chemotherapy Treated Triple Negative Breast Tumors. Front. Immunol. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Comen, E.A. Tracking the seed and tending the soil: Evolving concepts in metastatic breast cancer. Discov. Med. 2012, 14, 97–104. [Google Scholar] [PubMed]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.B.; Patwardhan, G.A.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390. [Google Scholar] [CrossRef]
- Berndt, B.; Zanker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef]
- Mortensen, K.; Lichtenberg, J.; Thomsen, P.D.; Larsson, L.-I. Spontaneous fusion between cancer cells and endothelial cells. Cell. Mol. Life Sci. 2004, 61, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Midtbö, K.; Andersson, H.; Åkerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindström, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. [Google Scholar] [CrossRef]
- Shabo, I.; Olsson, H.; Stål, O.; Svanvik, J. Breast Cancer Expression of DAP12 is Associated with Skeletal and Liver Metastases and Poor Survival. Clin. Breast Cancer 2013, 13, 371–377. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Cimas, F.J.; Manzano, A.; Baliu-Piqué, M.; García-Gil, E.; Pérez-Segura, P.; Nagy, Á.; Pandiella, A.; Győrffy, B.; Ocana, A. Genomic Mapping Identifies Mutations in RYR2 and AHNAK as Associated with Favorable Outcome in Basal-Like Breast Tumors Expressing PD1/PD-L. Cancers 2020, 12, 2243. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161.e5. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bullock, K.; Gurjao, C.; Braun, D.; Shukla, S.A.; Bossé, D.; Lalani, A.-K.A.; Gopal, S.; Jin, C.; Horak, C.; et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 2019, 10, 4346. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- FDA U.S. Food and Drug Administration. FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm560167.htm (accessed on 10 September 2020).
- Looney, A.-M.; Nawaz, K.; Webster, R.M. Tumour-agnostic therapies. Nat. Rev. Drug Discov. 2020, 19, 383–384. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Drescher, K.M.; Sharma, P.; Watson, P.; Gatalica, Z.; Thibodeau, S.N.; Lynch, H.T. Lymphocyte recruitment into the tumor site is altered in patients with MSI-H colon cancer. Fam. Cancer 2009, 8, 231–239. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.C.; Sen, S.; Hobbs, B.P.; Hong, D.S. Histology-agnostic drug development—considering issues beyond the tissue. Nat. Rev. Clin. Oncol. 2020, 17, 555–568. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Type of Alteration | Drug |
|---|---|---|
| ERBB2 | Amplification (DNA copy number ≥6; size ≤10 Mb) | Anti-HER2 monoclonal antibody (Trastuzumab) |
| BRCA 1/2 mutations | Germline mutations: Truncated mutations (InDel, splice-site, non- sense (except BRCA2 K3326X)) and rare known inactivating missense mutations | PARP inhibitor (Olaparib) |
| PIK3CA | Major hot-spot activating missense mutations | Alpha-specific PI3K inhibitor (Alpelisib) |
| NTRK | Translocations | Pan-Trk inhibitor (Larotrectinib) |
| - | Microsatellite instability (MSI) | Anti-PD1 antibody (Pembrolizumab) |
| Gene | Type of Alteration | Drug | Development Phase |
|---|---|---|---|
| Microsatellite instability–High (MSI-H) | Pembrolizumab | FDA approved | |
| NTRK | Gene fusions | Larotrectinib, Entrectinib | FDA approved |
| RET | Gene fusions/mutations | Selpercatinib Pralsetinib RXDX-105 TPX-0046 | Phase I/II Phase I/II Phase I/Ib Phase I/II |
| NRG1 | Gene fusions | Zenocutuzumab Tarloxotinib | Phase I/II basket |
| FGFR (fibroblast growth factor receptor) | Gene fusions/mutations | Debio 1347 TAS-120 | Phase II basket |
| KRASG12C | Mutations | AMG 510 MRTX849 | Phase I |
| TRK, ROS1, ALK | Mutations | Repotrectinib | Phase I/II |
| BRAF | Mutations | PLX8394 | Phase I/II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baliu-Piqué, M.; Pandiella, A.; Ocana, A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers 2020, 12, 3271. https://doi.org/10.3390/cancers12113271
Baliu-Piqué M, Pandiella A, Ocana A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers. 2020; 12(11):3271. https://doi.org/10.3390/cancers12113271
Chicago/Turabian StyleBaliu-Piqué, Mariona, Atanasio Pandiella, and Alberto Ocana. 2020. "Breast Cancer Heterogeneity and Response to Novel Therapeutics" Cancers 12, no. 11: 3271. https://doi.org/10.3390/cancers12113271
APA StyleBaliu-Piqué, M., Pandiella, A., & Ocana, A. (2020). Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers, 12(11), 3271. https://doi.org/10.3390/cancers12113271