Results and Clinical Interpretation of Germline RET Analysis in a Series of Patients with Medullary Thyroid Carcinoma: The Challenge of the Variants of Uncertain Significance

 , , ,
, , ,  ,
,  ,
, 
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics and Genetic Test Results
2.2. RET Test Results and Clinical Correlations
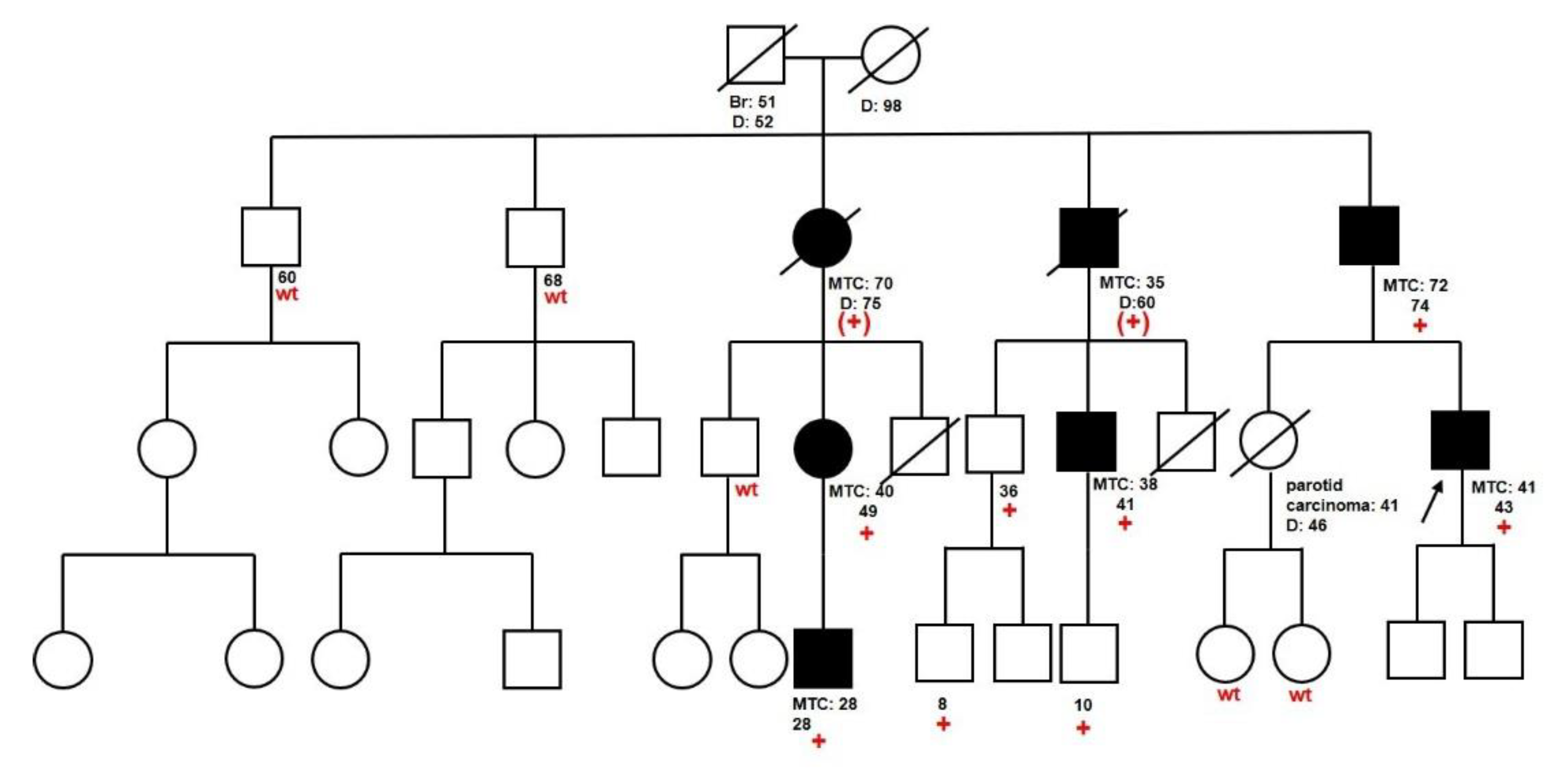
2.3. Families with Unclassified Variants
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Clinical Data
4.3. RET Analysis
4.4. Interpretation of Unclassified Variants
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Omur:, O.; Baran, Y. An update on molecular biology of thyroid cancers. Crit. Rev. Oncol. Hematol. 2014, 90, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6292–6306. [Google Scholar] [CrossRef] [PubMed]
- Salehian, B.; Samoa, R. RET gene abnormalities and thyroid disease: Who should be screened and when. J. Clin. Res. Pediatr. Endocrinol. 2013, 5 (Suppl. 1), 70–78. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Agrawal, V.; Pani, K.C.; Verma, R.; Jaiswal, S.; Mishra, A.; Pandey, R. C-cell hyperplasia in sporadic and familial medullary thyroid carcinoma. Indian J. Pathol. Microbiol. 2018, 61, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Lin, R.; Sosa, J.A. Prognosis of MTC: Demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 2006, 107, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Figlioli, G.; Landi, S.; Romei, C.; Elisei, R.; Gemignani, F. MTC (MTC) and RET proto-oncogene: Mutation spectrum in the familial cases and a meta-analysis of studies on the sporadic form. Mutat Res. 2013, 752, 36–44. [Google Scholar] [CrossRef]
- Raue, F.; Frank-Raue, K. Das medulläre Schilddrüsenkarzinom und die multiple endokrine Neoplasie Typ 2 [MTC and multiple endocrine neoplasia type 2]. Dtsch Med. Wochenschr. 2020, 10. [Google Scholar] [CrossRef]
- Wohllk, N.; Schweizer, H.; Erlic, Z.; Schmid, K.W.; Walz, M.K.; Raue, F.; Neumann, H.P. Multiple endocrine neoplasia type 2. Best Pr. Res. Clin. Endocrinol. Metab. 2010, 24, 371–387. [Google Scholar] [CrossRef]
- Moline, J.; Eng, C. Multiple endocrine neoplasia type 2: An overview. Genet. Med. 2011, 13, 755–764. [Google Scholar] [CrossRef]
- Howe, J.R.; Norton, J.A.; Wells, S.A., Jr. Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: Results of long-term follow-up. Surgery 1993, 114, 1070–1077. [Google Scholar]
- Larsen, L.V.; Mirebeau-Prunier, D.; Imai, T.; Alvarez-Escola, C.; Hasse-Lazar, K.; Censi, S.; Castroneves, L.A.; Sakurai, A.; Kihara, M.; Horiuchi, K.; et al. Primary hyperparathyroidism as first manifestation in multiple endocrine neoplasia type 2A: An international multicenter study. Endocr. Connect. 2020, 9, 489–497. [Google Scholar] [CrossRef]
- Brauckhoff, M.; Machens, A.; Hess, S.; Lorenz, K.; Gimm, O.; Brauckhoff, K.; Sekulla, C.; Dralle, H. Premonitory symptoms preceding metastatic medullary thyroid cancer in MEN 2B: An exploratory analysis. Surgery 2008, 144, 1044–1053. [Google Scholar] [CrossRef]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A., Jr. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993, 2, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; van Amstel, H.K.; Lips, C.J.; Nishisho, I.; Takai, S.I.; et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef]
- Yip, L.; Cote, G.J.; Shapiro, S.E.; Ayers, G.D.; Herzog, C.E.; Sellin, R.V.; Sherman, S.I.; Gagel, R.F.; Lee, J.E.; Evans, D.B. Multiple endocrine neoplasia type 2: Evaluation of the genotype-phenotype relationship. Arch. Surg. 2003, 138, 409–416. [Google Scholar] [CrossRef]
- Hedayati, M.; Zarif Yeganeh, M.; Sheikholeslami, S.; Afsari, F. Diversity of mutations in the RET proto-oncogene and its oncogenic mechanism in medullary thyroid cancer. Crit. Rev. Clin. Lab. Sci. 2016, 53, 217–227. [Google Scholar] [CrossRef]
- Raue, F.; Frank-Raue, K. Update on Multiple Endocrine Neoplasia Type 2: Focus on MTC. J. Endocr. Soc. 2018, 2, 933–943. [Google Scholar] [CrossRef]
- Voss, R.K.; Feng, L.; Lee, J.E.; Perrier, N.D.; Graham, P.H.; Hyde, S.M.; Nieves-Munoz, F.; Cabanillas, M.E.; Waguespack, S.G.; Cote, G.J.; et al. MTC in MEN2A: ATA Moderate- or High-Risk RET Mutations Do Not Predict Disease Aggressiveness. J. Clin. Endocrinol. Metab. 2017, 102, 2807–2813. [Google Scholar] [CrossRef]
- Kloos, R.T.; Eng, C.; Evans, D.B.; Francis, G.L.; Gagel, R.F.; Gharib, H.; Moley, J.F.; Pacini, F.; Ringel, M.D.; Schlumberger, M.; et al. Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid 2009, 19, 565–612. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of MTC. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Machens, A.; Lorenz, K.; Weber, F.; Dralle, H. Genotype-specific progression of hereditary medullary thyroid cancer. Hum. Mutat. 2018, 39, 860–869. [Google Scholar] [CrossRef]
- Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes 2019, 10, 698. [Google Scholar] [CrossRef]
- Cosci, B.; Vivaldi, A.; Romei, C.; Gemignani, F.; Landi, S.; Ciampi, R.; Tacito, A.; Molinaro, E.; Agate, L.; Bottici, V.; et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr. Relat. Cancer 2011, 18, 603–612. [Google Scholar] [CrossRef]
- Seri, M.; Yin, L.; Barone, V.; Bolino, A.; Celli, I.; Bocciardi, R.; Pasini, B.; Ceccherini, I.; Lerone, M.; Kristoffersson, U.; et al. Frequency of RET mutations in long- and short-segment Hirschsprung disease. Hum. Mutat. 1997, 9, 243–249. [Google Scholar] [CrossRef]
- Berndt, I.; Reuter, M.; Saller, B.; Frank-Raue, K.; Growth, P.; Grussendorf, M.; Raue, F.; Ritter, M.M.; Höppner, W. A new hot spot for mutations in the ret protooncogene causing familial MTC and multiple endocrine neoplasia type 2A. J. Clin. Endocrinol. Metab. 1998, 83, 770–774. [Google Scholar] [CrossRef]
- Tamanaha, R.; Camacho, C.P.; Ikejiri, E.S.; Maciel, R.M.; Cerutti, J.M. Y791F RET mutation and early onset of MTC in a Brazilian kindred: Evaluation of phenotype-modifying effect of germline variants. Clin. Endocrinol. 2007, 67, 806–808. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Vierhapper, H.; Bieglmayer, C.; Heinze, G.; Baumgartner-Parzer, S. Frequency of RET proto-oncogene mutations in patients with normal and with moderately elevated pentagastrin-stimulated serum concentrations of calcitonin. Thyroid 2004, 14, 580–583. [Google Scholar] [CrossRef]
- Toledo, R.A.; Loureço, D.M., Jr.; Camacho, C.; Lindsey, S.; Cerutti, J.; Maciel, R.M.; Toledo, S.P. RET Y791F: Alone or accompanied? Arch. Endocrinol. Metab. 2015, 59, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Dorschner, M.O.; Robertson, P.D.; Salama, J.S.; Hart, R.; Shirts, B.H.; Murray, M.L.; Tokita, M.J.; Gallego, C.J.; Kim, D.S.; et al. Actionable exomic incidental findings in 6503 participants: Challenges of variant classification. Genome Res. 2015, 25, 305–315. [Google Scholar] [CrossRef]
- Toledo, R.A.; Hatakana, R.; Lourenço, D.M., Jr.; Lindsey, S.C.; Camacho, C.P.; Almeida, M.; Lima, J.V., Jr.; Sekiya, T.; Garralda, E.; Naslavsky, M.S.; et al. Comprehensive assessment of the disputed RET Y791F variant shows no association with MTC susceptibility. Endocr. Relat. Cancer 2015, 22, 65–76. [Google Scholar] [CrossRef]
- Vestergaard, P.; Vestergaard, E.M.; Brockstedt, H.; Christiansen, P. Codon Y791F mutations in a large kindred: Is prophylactic thyroidectomy always indicated? World J. Surg. 2007, 31, 996–1004. [Google Scholar] [CrossRef]
- Erlic, Z.; Hoffmann, M.M.; Sullivan, M.; Franke, G.; Peczkowska, M.; Harsch, I.; Schott, M.; Gabbert, H.E.; Valimäki, M.; Preuss, S.F.; et al. Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 308–313. [Google Scholar] [CrossRef]
- Toledo, R.A.; Wagner, S.M.; Coutinho, F.L.; Lourenço, D.M., Jr.; Azevedo, J.A.; Longuini, V.C.; Reis, M.T.; Siqueira, S.A.; Lucon, A.M.; Tavares, M.R.; et al. High penetrance of pheochromocytoma associated with the novel C634Y/Y791F double germline mutation in the RET protooncogene. J. Clin. Endocrinol. Metab. 2010, 95, 1318–1327. [Google Scholar] [CrossRef]
- Pęczkowska, M.; Kowalska, A.; Sygut, J.; Waligórski, D.; Malinoc, A.; Janaszek-Sitkowska, H.; Prejbisz, A.; Januszewicz, A.; Neumann, H.P. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin. Endocrinol. 2013, 79, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Colombo-Benkmann, M.; Li, Z.; Riemann, B.; Hengst, K.; Herbst, H.; Keuser, R.; Gross, U.; Rondot, S.; Raue, F.; Senninger, N.; et al. Characterization of the RET protooncogene transmembrane domain mutation S649L associated with nonaggressive medullary thyroid carcinoma. Eur. J. Endocrinol. 2008, 158, 811–816. [Google Scholar] [CrossRef]
- Latteyer, S.; Klein-Hitpass, L.; Khandanpour, C.; Zwanziger, D.; Poeppel, T.D.; Schmid, K.W.; Führer, D.; Moeller, L.C. A 6-Base Pair in Frame Germline Deletion in Exon 7 of RET Leads to Increased RET Phosphorylation, ERK Activation, and MEN2A. J. Clin. Endocrinol. Metab. 2016, 101, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, I.; Vigano, E.; Alberti, L.; Mondellini, P.; Uggeri, M.; Pasini, B.; Borrello, M.G.; Pierotti, M.A. The Glu632-Leu633 deletion in cysteine rich domain of Ret induces constitutive dimerization and alters the processing of the receptor protein. Oncogene 1999, 18, 4833–4838. [Google Scholar] [CrossRef]
- Ceccherini, I.; Pasini, B.; Pacini, F.; Gullo, M.; Bongarzone, I.; Romei, C.; Santamaria, G.; Matera, I.; Mondellini, P.; Scopsi, L.; et al. Somatic in frame deletions not involving juxtamembranous cysteine residues strongly activate the RET proto-oncogene. Oncogene 1997, 14, 2609–2612. [Google Scholar] [CrossRef]
- Mulligan, L.M. 65 YEARS OF THE DOUBLE HELIX: Exploiting insights on the RET receptor for personalized cancer medicine. Endocr. Relat. Cancer 2018, 25, T189–T200. [Google Scholar] [CrossRef] [PubMed]
- Krampitz, G.W.; Norton, J.A. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial MTC. Cancer 2014, 120, 1920–1931. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Cosci, B.; Agate, L.; Bottici, V.; Molinaro, E.; Sculli, M.; Miccoli, P.; Basolo, F.; Grasso, L.; et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: Experience with 807 individuals at one center. J. Clin. Endocrinol. Metab. 2007, 92, 4725–4729. [Google Scholar] [CrossRef]
- Bugalho, M.J.; Domingues, R.; Santos, J.R.; Catarino, A.L.; Sobrinho, L. Mutation analysis of the RET proto-oncogene and early thyroidectomy: Results of a Portuguese cancer centre. Surgery 2007, 141, 90–95. [Google Scholar] [CrossRef]
- Wiench, M.; Wygoda, Z.; Gubala, E.; Wloch, J.; Lisowska, K.; Krassowski, J.; Scieglinska, D.; Fiszer-Kierzkowska, A.; Lange, D.; Kula, D.; et al. Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. J. Clin. Oncol. 2001, 19, 1374–1380. [Google Scholar] [CrossRef]
- Romei, C.; Cosci, B.; Renzini, G.; Bottici, V.; Molinaro, E.; Agate, L.; Passannanti, P.; Viola, D.; Biagini, A.; Basolo, F.; et al. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin. Endocrinol. 2011, 74, 241–247. [Google Scholar] [CrossRef]
- Mulligan, L.M.; Marsh, D.J.; Robinson, B.G.; Schuffenecker, I.; Zedenius, J.; Lips, C.J.; Gagel, R.F.; Takai, S.I.; Noll, W.W.; Fink, M.; et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: Report of the International RET Mutation Consortium. J. Intern. Med. 1995, 238, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Gimm, O.; Marsh, D.J.; Andrew, S.D.; Frilling, A.; Dahia, P.L.; Mulligan, L.M.; Zajac, J.D.; Robinson, B.G.; Eng, C. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J. Clin. Endocrinol. Metab. 1997, 82, 3902–3904. [Google Scholar] [CrossRef]
- Iwashita, T.; Murakami, H.; Kurokawa, K.; Kawai, K.; Miyauchi, A.; Futami, H.; Qiao, S.; Ichihara, M.; Takahashi, M. A two-hit model for development of multiple endocrine neoplasia type 2B by RET mutations. Biochem. Biophys. Res. Commun. 2000, 268, 804–808. [Google Scholar] [CrossRef]
- Miyauchi, A.; Futami, H.; Hai, N.; Yokozawa, T.; Kuma, K.; Aoki, N.; Kosugi, S.; Sugano, K.; Yamaguchi, K. Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn. J. Cancer Res. 1999, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bihan, H.; Murat, A.; Fysekidis, M.; Al-Salameh, A.; Schwartz, C.; Baudin, E.; Thieblot, P.; Borson-Chazot, F.; Guillausseau, P.J.; Cardot-Bauters, C.; et al. The clinical spectrum of RET proto-oncogene mutations in codon 790. Eur. J. Endocrinol. 2013, 169, 271–276. [Google Scholar] [CrossRef]
- Margraf, R.L.; Crockett, D.K.; Krautscheid, P.M.; Seamons, R.; Calderon, F.R.; Wittwer, C.T.; Mao, R. Multiple endocrine neoplasia type 2 RET protooncogene database: Repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum. Mutat. 2009, 30, 548–556. [Google Scholar] [CrossRef]


{kind=link}
| Nucleotide Variant | Aminoacidic Change | ATA Risk Level | N. Families | N. Carriers | MTC/tot Carriers a (%) | PCC/Tot Carriers a (%) | PHPT/Tot Carriers a (%) | MEN2B Manifestations b/Tot Carriers a (%) |
|---|---|---|---|---|---|---|---|---|
| c.2671T > G | p.Ser891Ala | MOD | 1 | 1 | 0/1 (0.0) | 0/1 (0.0) | 0/1 (0.0) | 0/1 (0.0) |
| c.2410G > A | p.Val804Met | MOD | 5 | 14 | 6/12 (50.0) | 0/12 (0.0) | 0/12 (0.0) | 0/12 (0.0) |
| c.2370G > T | p.Leu790Phe | MOD | 1 | 9 | 7/9 (77.8) | 1/9 (11.1) | 0/9 (0.0) | 0/9 (0.0) |
| c. 2304G > C | p.Glu768Asp | MOD | 2 | 10 | 3/10 (30.0) | 0/10 (0.0) | 0/10 (0.0) | 0/10 (0.0) |
| c.1901G > T | p.Cys634Phe | H | 1 | 1 | 1/1 (100.0) | 1/1 (100.0) | 0/1 (0.0) | 0/1 (0.0) |
| c.1901G > A | p.Cys634Tyr | H | 0 | 1 | 1/1 (100.0) | 1/1 (100.0) | 0/1 (0.0) | 0/1 (0.0) |
| c.2753T > C | p.Met918Thr | HST | 2 | 2 | 2/2 (100.0) | 0/1 (0.0) | 0/1 (0.0) | 1/1 (100.0) |
| Feature | RET VARIANT DETECTED | p Value | |||||
|---|---|---|---|---|---|---|---|
| NONE | ALL | MOD | HST/H | (NONE vs. ALL) | (NONE vs. MOD) | (MOD vs. HST/H) | |
| Age at diagnosis of MTC (mean) | 56.42 | 44.45 | 51.78 | 11.50 | 0.010 | 0.281 | <0.001 |
| Sex, n. Female/tot (%) | 32/48 (66.7) | 13/22 (59.1) | 10/18 (55.6) | 3/4 (75.0) | 0.597 | 0.408 | 0.616 |
| Presence of other tumors, n./tot (%) | 14/48 (29.2) | 6/22 (27.3) | 4/18 (22.2) | 2/4 (50.0) | 1.000 | 0.759 | 0.292 |
| Positive family history, n./tot (%) | 1/48 (2.1) | 15/22 (68.2) | 13/18 (72.2) | 2/4 (50.0) | <0.001 | <0.001 | 0.565 |
| Stage, n. T > 1/tota (%) | 8/22 (36.4) | 3/7 (42.9) | 2/6 (33.3) | 1/1 (100.0) | 1.000 | 1.000 | 0.429 |
| Nucleotide Variant | Aminoacidic Change | Allele Frequency a | Mean Conservation Score b | Clinvar Class | Varsome Class (Computational Verdicts) | N. Carriers (n. Affected by MTC) |
|---|---|---|---|---|---|---|
| c.2711C > T | p.Ser904Phe | NP | 5.5799 | LP | LP (10 D vs. 1 B) | 8 (5) |
| c.2372A > T | p.Tyr791Phe | 0.00209 | 5.34 | CI | LB (8 D vs. 3 B) | 1 (1) |
| c.2129A > G | p.Lys710Arg | 0.00000816 | 3.72 | US | US (8 D vs. 3 B) | 1 (0) |
| c.1946C > T | p.Ser649Leu | 0.0003164 | 4.34 | CI | LP (11 D vs. 0 B) | 1 (1) |
| c.1893_1898delCGAGCT | p.Asp631_Leu633delinsGlu | NP | 1.2967 | NP | LP (1 D vs. 0 B) | 1 (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Innella, G.; Rossi, C.; Romagnoli, M.; Repaci, A.; Bianchi, D.; Cantarini, M.E.; Martorana, D.; Godino, L.; Pession, A.; Percesepe, A.; et al. Results and Clinical Interpretation of Germline RET Analysis in a Series of Patients with Medullary Thyroid Carcinoma: The Challenge of the Variants of Uncertain Significance. Cancers 2020, 12, 3268. https://doi.org/10.3390/cancers12113268
Innella G, Rossi C, Romagnoli M, Repaci A, Bianchi D, Cantarini ME, Martorana D, Godino L, Pession A, Percesepe A, et al. Results and Clinical Interpretation of Germline RET Analysis in a Series of Patients with Medullary Thyroid Carcinoma: The Challenge of the Variants of Uncertain Significance. Cancers. 2020; 12(11):3268. https://doi.org/10.3390/cancers12113268
Chicago/Turabian StyleInnella, Giovanni, Cesare Rossi, Maria Romagnoli, Andrea Repaci, Davide Bianchi, Maria Elena Cantarini, Davide Martorana, Lea Godino, Andrea Pession, Antonio Percesepe, and et al. 2020. "Results and Clinical Interpretation of Germline RET Analysis in a Series of Patients with Medullary Thyroid Carcinoma: The Challenge of the Variants of Uncertain Significance" Cancers 12, no. 11: 3268. https://doi.org/10.3390/cancers12113268
APA StyleInnella, G., Rossi, C., Romagnoli, M., Repaci, A., Bianchi, D., Cantarini, M. E., Martorana, D., Godino, L., Pession, A., Percesepe, A., Pagotto, U., & Turchetti, D. (2020). Results and Clinical Interpretation of Germline RET Analysis in a Series of Patients with Medullary Thyroid Carcinoma: The Challenge of the Variants of Uncertain Significance. Cancers, 12(11), 3268. https://doi.org/10.3390/cancers12113268