IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Mice
2.2. Hematopoietic Stem Cell Transplantation
2.3. Flow Cytometry
2.4. Cytotoxic Assays
2.5. Serum Cytokine Bead Array
2.6. In Vivo Tumor Models
2.7. Statistical Analysis
3. Results
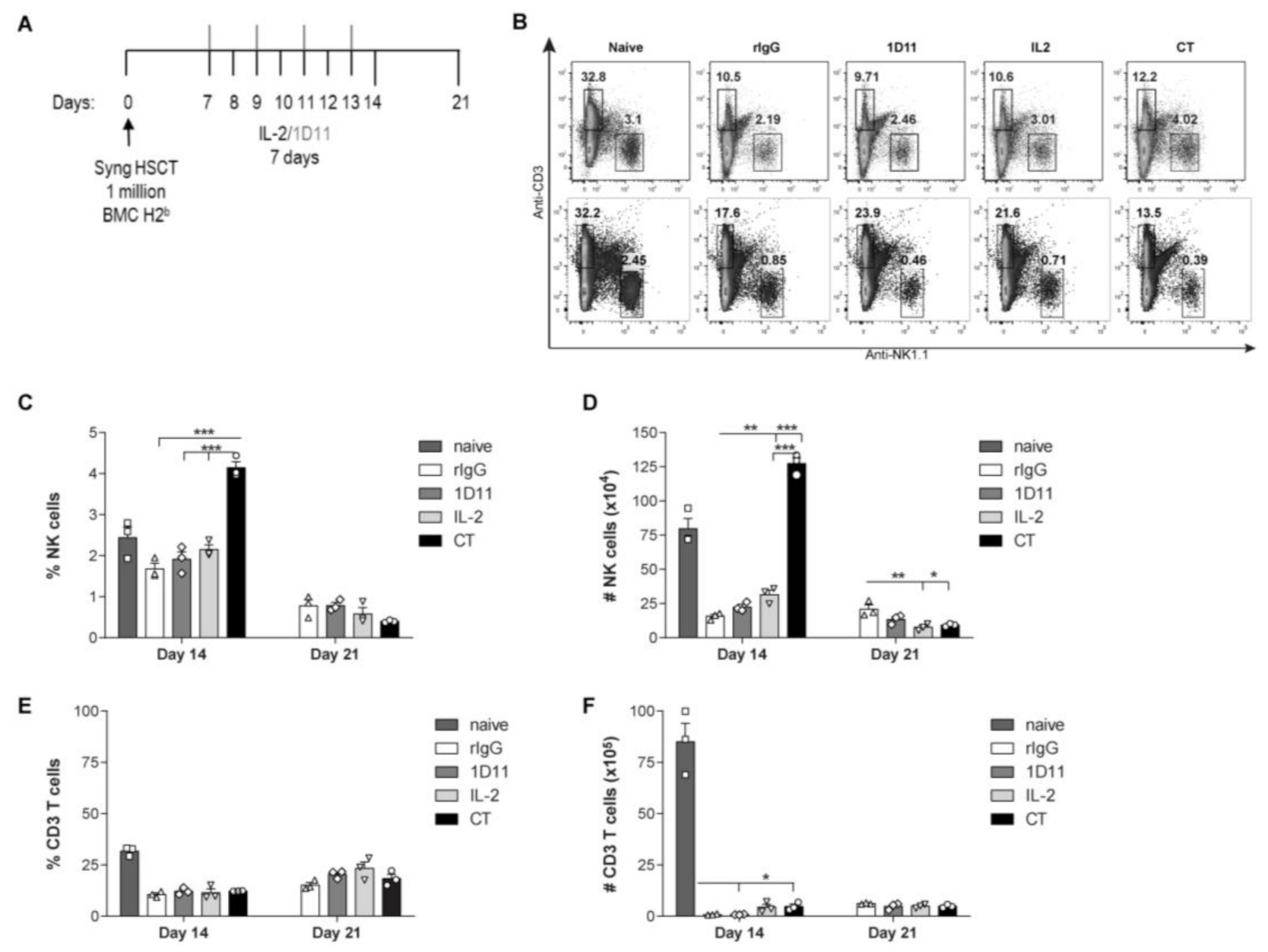
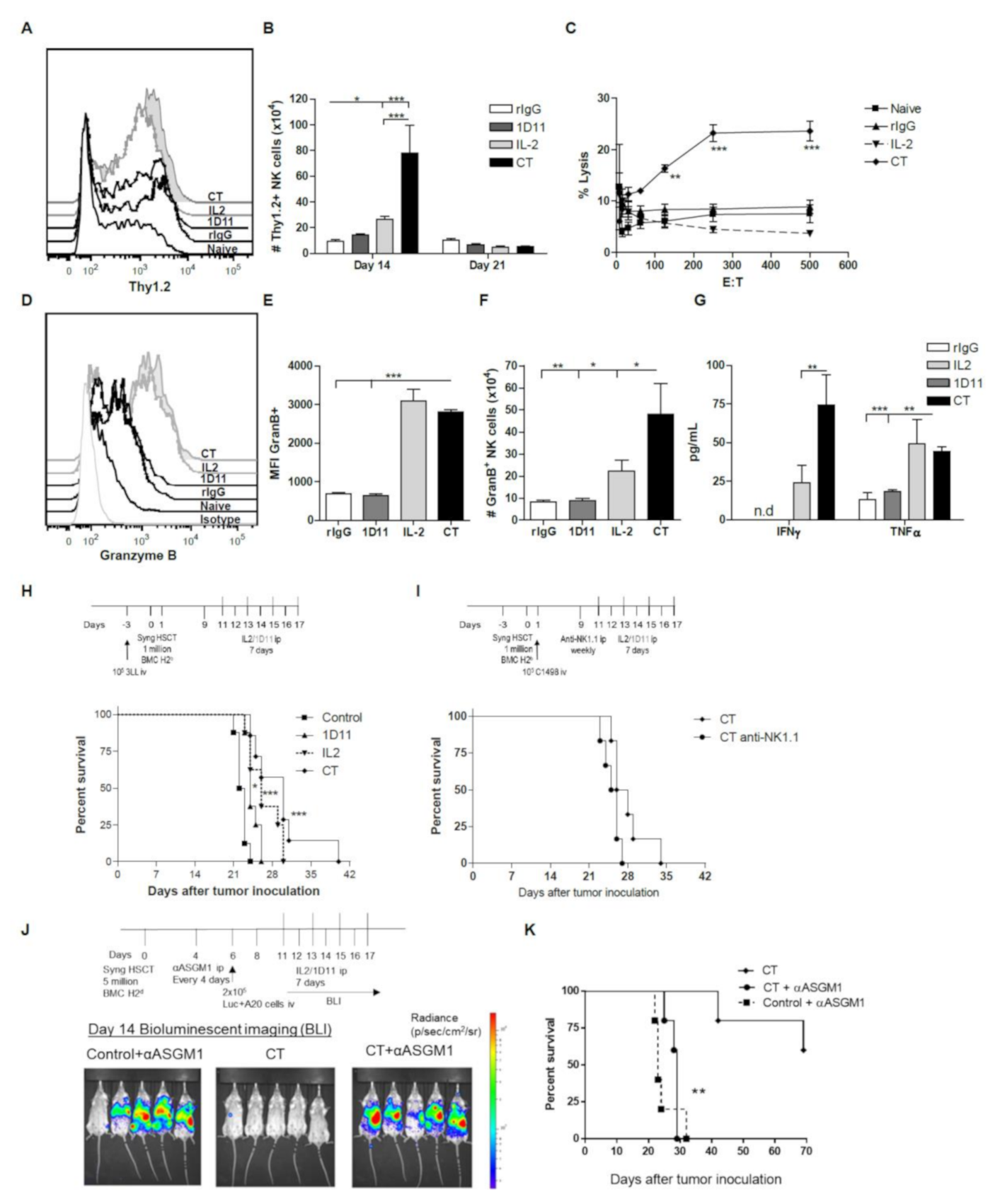
3.1. IL-2 and Anti-TGF-β Combination Therapy (CT) Results in Marked NK Cell Expansion after Congenic HSCT
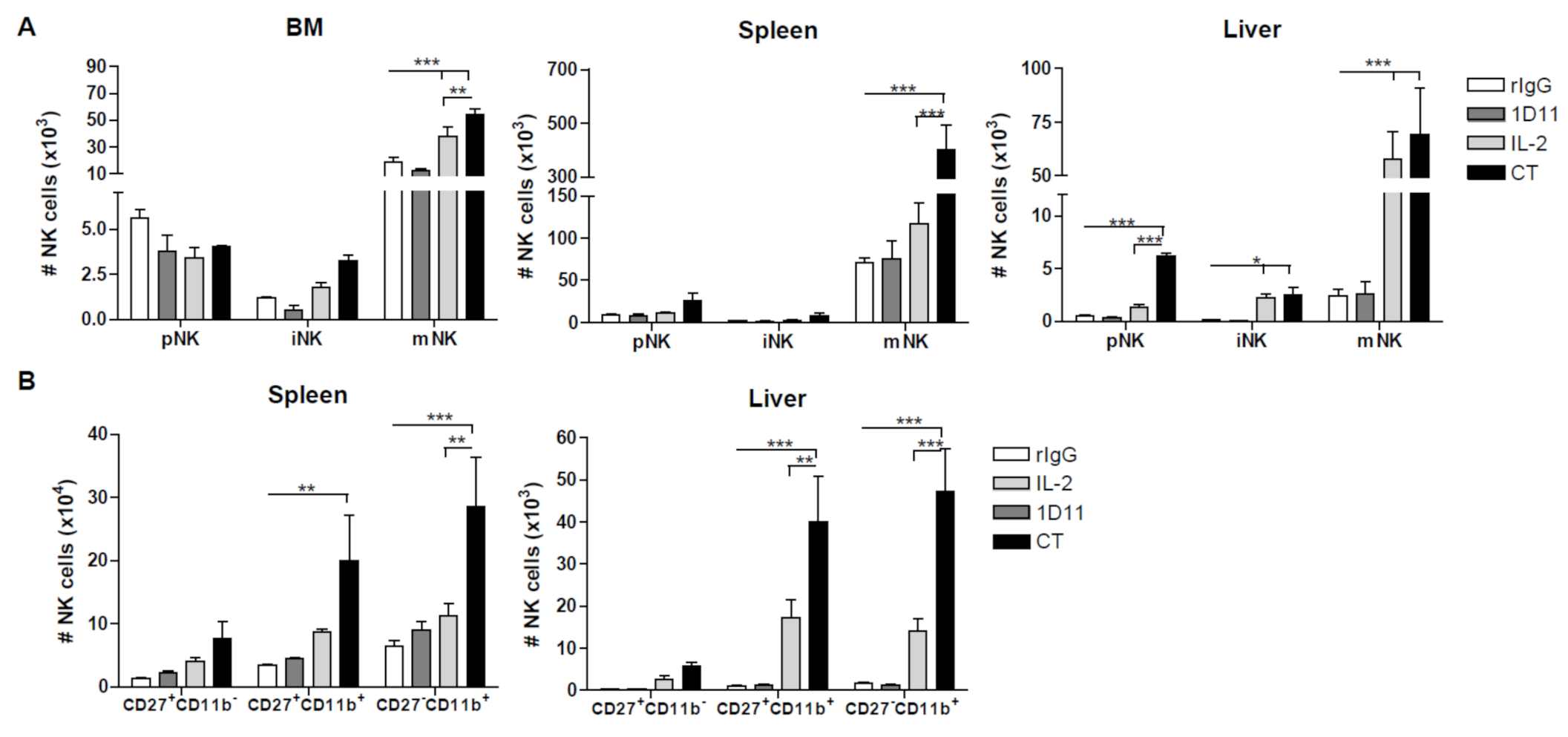
3.2. Administration of IL-2 Combined with Anti-TGF-β Accelerates NK Cell Reconstitution after HSCT
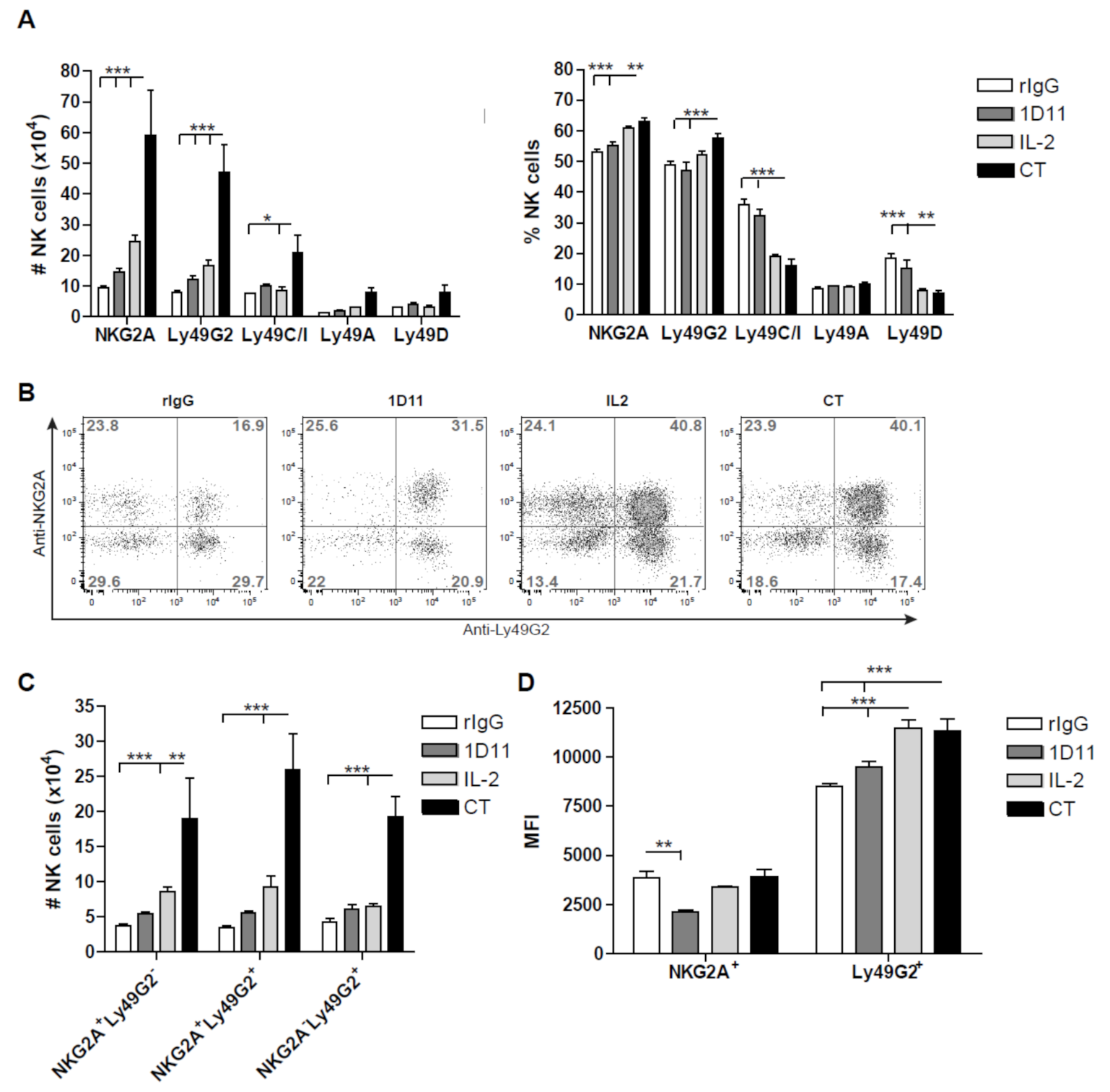
3.3. Early Recovery of NK Cells Co-expressing the Inhibitory Receptors NKG2A and Ly49G2 is Observed after NK Cell Stimulation during HSCT
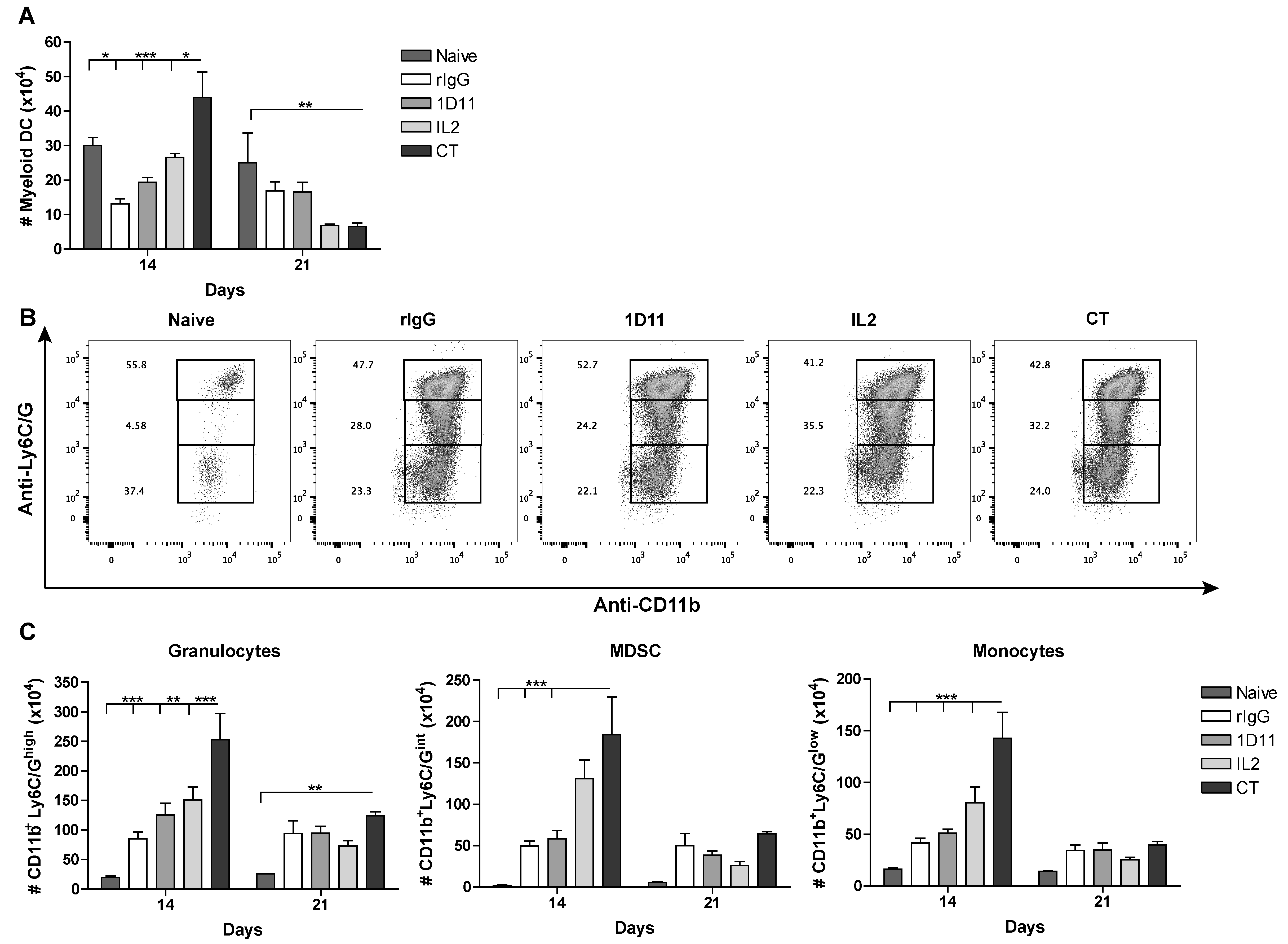
3.4. CT Improves Reconstitution of Myeloid-derived Cells after HSCT
3.5. Influence of CT on the NK cell Functional Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 17, 475–492. [Google Scholar] [CrossRef]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef]
- Pierini, A.; Alvarez, M.; Negrin, R.S. NK Cell and CD4+FoxP3+ Regulatory T Cell Based Therapies for Hematopoietic Stem Cell Engraftment. Stem Cells Int. 2016, 2016, 9025835. [Google Scholar] [CrossRef]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsis-Mortari, A.; Keever-Taylor, C.A.; Zhang, M.J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow Transplant. 2003, 32, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Alici, E. Natural killer cell-based immunotherapy in cancer: Current insights and future prospects. J. Intern. Med. 2009, 266, 154–181. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef]
- Colucci, F.; Caligiuri, M.A.; Di Santo, J.P. What does it take to make a natural killer? Nat. Rev. Immunol. 2003, 3, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Sun, K.; Murphy, W.J. Mouse host unlicensed NK cells promote donor allogeneic bone marrow engraftment. Blood 2016, 127, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, J.P. A defining factor for natural killer cell development. Nat. Immunol. 2009, 10, 1051–1052. [Google Scholar] [CrossRef]
- Cooper, M.A.; Bush, J.E.; Fehniger, T.A.; VanDeusen, J.B.; Waite, R.E.; Liu, Y.; Aguila, H.L.; Caligiuri, M.A. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood 2002, 100, 3633–3638. [Google Scholar] [CrossRef]
- Alvarez, M.; Ochoa, M.C.; Minute, L.; Melero, I.; Berraondo, P. Rapid isolation and enrichment of mouse NK cells for experimental purposes. Methods Enzymol. 2020, 631, 257–275. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Caligiuri, M.A. Interleukin-2 and interleukin-15: Immunotherapy for cancer. Cytokine Growth Factor Rev. 2002, 13, 169–183. [Google Scholar] [CrossRef]
- Hallett, W.H.; Ames, E.; Alvarez, M.; Barao, I.; Taylor, P.A.; Blazar, B.R.; Murphy, W.J. Combination therapy using IL-2 and anti-CD25 results in augmented natural killer cell-mediated antitumor responses. Biol. Blood Marrow Transpl. 2008, 14, 1088–1099. [Google Scholar] [CrossRef]
- Boyman, O.; Surh, C.D.; Sprent, J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin. Biol. Ther. 2006, 6, 1323–1331. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Ksendzovsky, A.; Feinstein, D.; Zengou, R.; Sharp, A.; Polak, P.; Lichtor, T.; Glick, R.P. Investigation of immunosuppressive mechanisms in a mouse glioma model. J. Neuro-Oncol. 2009, 93, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Penafuerte, C.; Galipeau, J. TGF beta secreted by B16 melanoma antagonizes cancer gene immunotherapy bystander effect. Cancer Immunol. Immunother. 2008, 57, 1197–1206. [Google Scholar] [CrossRef]
- Rook, A.H.; Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Sporn, M.B.; Burlington, D.B.; Lane, H.C.; Fauci, A.S. Effects of transforming growth factor beta on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986, 136, 3916–3920. [Google Scholar]
- Bellone, G.; Aste-Amezaga, M.; Trinchieri, G.; Rodeck, U. Regulation of NK cell functions by TGF-beta 1. J. Immunol. 1995, 155, 1066–1073. [Google Scholar] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGF-beta. Nat. Rev. 2010, 10, 554–567. [Google Scholar] [CrossRef]
- Petrausch, U.; Jensen, S.M.; Twitty, C.; Poehlein, C.H.; Haley, D.P.; Walker, E.B.; Fox, B.A. Disruption of TGF-beta signaling prevents the generation of tumor-sensitized regulatory T cells and facilitates therapeutic antitumor immunity. J. Immunol. 2009, 183, 3682–3689. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.S.; Terabe, M.; Mamura, M.; Kang, M.J.; Chae, H.; Stuelten, C.; Kohn, E.; Tang, B.; Sabzevari, H.; Anver, M.R.; et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008, 68, 3835–3843. [Google Scholar] [CrossRef] [PubMed]
- Perry, K.; Wong, L.; Liu, V.; Park, I.; Zhang, Q.; Rejen, V.; Huang, X.; Smith, N.D.; Jovanovic, B.; Lonning, S.; et al. Treatment of transforming growth factor-beta-insensitive mouse Renca tumor by transforming growth factor-beta elimination. Urology 2008, 72, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Shah, A.H.; Tabayoyong, W.B.; Kimm, S.Y.; Kim, S.J.; Van Parijs, L.; Lee, C. Reconstitution of lethally irradiated adult mice with dominant negative TGF-beta type II receptor-transduced bone marrow leads to myeloid expansion and inflammatory disease. J. Immunol. 2002, 169, 3485–3491. [Google Scholar] [CrossRef]
- Ueda, R.; Fujita, M.; Zhu, X.; Sasaki, K.; Kastenhuber, E.R.; Kohanbash, G.; McDonald, H.A.; Harper, J.; Lonning, S.; Okada, H. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin. Cancer Res. 2009, 15, 6551–6559. [Google Scholar] [CrossRef]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- Alvarez, M.; Bouchlaka, M.N.; Sckisel, G.D.; Sungur, C.M.; Chen, M.; Murphy, W.J. Increased Antitumor Effects Using IL-2 with Anti-TGF-beta Reveals Competition between Mouse NK and CD8 T Cells. J. Immunol. 2014. [Google Scholar] [CrossRef]
- Barao, I.; Alvarez, M.; Ames, E.; Orr, M.T.; Stefanski, H.E.; Blazar, B.R.; Lanier, L.L.; Anderson, S.K.; Redelman, D.; Murphy, W.J. Mouse Ly49G2+ NK cells dominate early responses during both immune reconstitution and activation independently of MHC. Blood 2011, 117, 7032–7041. [Google Scholar] [CrossRef]
- Alvarez, M.; Sungur, C.M.; Ames, E.; Anderson, S.K.; Pomeroy, C.; Murphy, W.J. Contrasting effects of anti-Ly49A due to MHC class I cis binding on NK cell-mediated allogeneic bone marrow cell resistance. J. Immunol. 2013, 191, 688–698. [Google Scholar] [CrossRef]
- Melder, R.J.; Osborn, B.L.; Riccobene, T.; Kanakaraj, P.; Wei, P.; Chen, G.; Stolow, D.; Halpern, W.G.; Migone, T.S.; Wang, Q.; et al. Pharmacokinetics and in vitro and in vivo anti-tumor response of an interleukin-2-human serum albumin fusion protein in mice. Cancer Immunol. Immunother. 2005, 54, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Li, X.; Jha, S.; Wang, W.; Karetskaya, L.; Pratt, B.; Ledbetter, S. Therapeutic role of TGF-beta-neutralizing antibody in mouse cyclosporin A nephropathy: Morphologic improvement associated with functional preservation. J. Am. Soc. Nephrol. 2003, 14, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S. Therapeutic applications: Natural killer cells in the clinic. Hematology/the Education Program of the American Society of Hematology. Am. Soc. Hematol. Educ. Prog. 2013, 2013, 247–253. [Google Scholar] [CrossRef]
- Marcoe, J.P.; Lim, J.R.; Schaubert, K.L.; Fodil-Cornu, N.; Matka, M.; McCubbrey, A.L.; Farr, A.R.; Vidal, S.M.; Laouar, Y. TGF-beta is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat. Immunol. 2012, 13, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Smyth, M.J. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 2006, 176, 1517–1524. [Google Scholar] [CrossRef]
- Watt, S.V.; Andrews, D.M.; Takeda, K.; Smyth, M.J.; Hayakawa, Y. IFN-gamma-dependent recruitment of mature CD27(high) NK cells to lymph nodes primed by dendritic cells. J. Immunol. 2008, 181, 5323–5330. [Google Scholar] [CrossRef]
- Nguyen, S.; Dhedin, N.; Vernant, J.P.; Kuentz, M.; Al Jijakli, A.; Rouas-Freiss, N.; Carosella, E.D.; Boudifa, A.; Debre, P.; Vieillard, V. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: Immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005, 105, 4135–4142. [Google Scholar] [CrossRef]
- Park, S.M.; Deering, R.P.; Lu, Y.; Tivnan, P.; Lianoglou, S.; Al-Shahrour, F.; Ebert, B.L.; Hacohen, N.; Leslie, C.; Daley, G.Q.; et al. Musashi-2 controls cell fate, lineage bias, and TGF-beta signaling in HSCs. J. Exp. Med. 2014, 211, 71–87. [Google Scholar] [CrossRef]
- Garbe, A.; Spyridonidis, A.; Mobest, D.; Schmoor, C.; Mertelsmann, R.; Henschler, R. Transforming growth factor-beta 1 delays formation of granulocyte-macrophage colony-forming cells, but spares more primitive progenitors during ex vivo expansion of CD34+ haemopoietic progenitor cells. Br. J. Haematol. 1997, 99, 951–958. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef]
- Wojtowicz-Praga, S.; Verma, U.N.; Wakefield, L.; Esteban, J.M.; Hartmann, D.; Mazumder, A. Modulation of B16 melanoma growth and metastasis by anti-transforming growth factor beta antibody and interleukin-2. J. Immunother. 1996, 19, 169–175. [Google Scholar] [CrossRef]
- Qin, W.; Tian, F.; Wang, F.; Song, B.; Wang, H.; Zhang, Q.; Jovanovic, B.; Liang, L.; Guo, Y.; Smith, N.; et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive cytolytic T cells for treatment of established mouse Renca tumors. Urology 2008, 72, 943–947. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef]
- Carli, C.; Giroux, M.; Delisle, J.S. Roles of transforming growth factor-beta in graft-versus-host and graft-versus-tumor effects. Biol. Blood Marrow Transpl. 2012, 18, 1329–1340. [Google Scholar] [CrossRef]
- Letourneau, S.; van Leeuwen, E.M.; Krieg, C.; Martin, C.; Pantaleo, G.; Sprent, J.; Surh, C.D.; Boyman, O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc. Natl. Acad. Sci. USA 2010, 107, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Simonetta, F.; Baker, J.; Pierini, A.; Wenokur, A.S.; Morrison, A.R.; Murphy, W.J.; Negrin, R.S. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Alvarez, M.; Simonetta, F.; Baker, J.; Morrison, A.R.; Wenokur, A.S.; Pierini, A.; Berraondo, P.; Negrin, R.S. Indirect Impact of PD-1/PD-L1 Blockade on a Murine Model of NK Cell Exhaustion. Front. Immunol. 2020, 11, 7. [Google Scholar] [CrossRef]
- Elpek, K.G.; Rubinstein, M.P.; Bellemare-Pelletier, A.; Goldrath, A.W.; Turley, S.J. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc. Natl. Acad. Sci. USA 2010, 107, 21647–21652. [Google Scholar] [CrossRef]
- Gill, S.; Vasey, A.E.; De Souza, A.; Baker, J.; Smith, A.T.; Kohrt, H.E.; Florek, M.; Gibbs, K.D., Jr.; Tate, K.; Ritchie, D.S.; et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood 2012, 119, 5758–5768. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Hanke, T.; Takizawa, H.; McMahon, C.W.; Busch, D.H.; Pamer, E.G.; Miller, J.D.; Altman, J.D.; Liu, Y.; Cado, D.; Lemonnier, F.A.; et al. Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity 1999, 11, 67–77. [Google Scholar] [CrossRef]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Alvarez, M.; Ames, E.; Barao, I.; Chen, M.; Longo, D.L.; Redelman, D.; Murphy, W.J. Mouse NK cell-mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood 2012, 119, 1590–1598. [Google Scholar] [CrossRef]
- Miller, J.S.; McCullar, V. Human natural killer cells with polyclonal lectin and immunoglobulinlike receptors develop from single hematopoietic stem cells with preferential expression of NKG2A and KIR2DL2/L3/S2. Blood 2001, 98, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Dulphy, N.; Haas, P.; Busson, M.; Belhadj, S.; Peffault de Latour, R.; Robin, M.; Carmagnat, M.; Loiseau, P.; Tamouza, R.; Scieux, C.; et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J. Immunol. 2008, 181, 2227–2237. [Google Scholar] [CrossRef]
- Bjorkstrom, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Bjorklund, A.T.; Flodstrom-Tullberg, M.; Michaelsson, J.; Rottenberg, M.E.; et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Verges, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- BitMansour, A.; Burns, S.M.; Traver, D.; Akashi, K.; Contag, C.H.; Weissman, I.L.; Brown, J.M. Myeloid progenitors protect against invasive aspergillosis and Pseudomonas aeruginosa infection following hematopoietic stem cell transplantation. Blood 2002, 100, 4660–4667. [Google Scholar] [CrossRef]
- Caraglia, M.; Correale, P.; Giannicola, R.; Staropoli, N.; Botta, C.; Pastina, P.; Nesci, A.; Caporlingua, N.; Francini, E.; Ridolfi, L.; et al. GOLFIG Chemo-Immunotherapy in Metastatic Colorectal Cancer Patients. A Critical Review on a Long-Lasting Follow-Up. Front. Oncol. 2019, 9, 1102. [Google Scholar] [CrossRef]
- Eigentler, T.K.; Weide, B.; de Braud, F.; Spitaleri, G.; Romanini, A.; Pflugfelder, A.; Gonzalez-Iglesias, R.; Tasciotti, A.; Giovannoni, L.; Schwager, K.; et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL-2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin. Cancer Res. 2011, 17, 7732–7742. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, M.; Spitaleri, G.; Curigliano, G.; Roigas, J.; Weikert, S.; Kempkensteffen, C.; Roemer, A.; Kloeters, C.; Rogalla, P.; Pecher, G.; et al. The tumour-targeting human L19-IL-2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur. J. Cancer 2010, 46, 2926–2935. [Google Scholar] [CrossRef]
- Pretto, F.; Elia, G.; Castioni, N.; Neri, D. Preclinical evaluation of IL-2-based immunocytokines supports their use in combination with dacarbazine, paclitaxel and TNF-based immunotherapy. Cancer Immunol. Immunother. 2014, 63, 901–910. [Google Scholar] [CrossRef]
- Schliemann, C.; Gutbrodt, K.L.; Kerkhoff, A.; Pohlen, M.; Wiebe, S.; Silling, G.; Angenendt, L.; Kessler, T.; Mesters, R.M.; Giovannoni, L.; et al. Targeting interleukin-2 to the bone marrow stroma for therapy of acute myeloid leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Cancer Immunol. Res. 2015, 3, 547–556. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, H.; Li, Y.; Zhang, R.; Kleeff, J.; Zhang, X.; Cui, M.; Liu, J.; Li, T.; Gao, J.; et al. Combined blockade of TGF-beta1 and GM-CSF improves chemotherapeutic effects for pancreatic cancer by modulating tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 1477–1492. [Google Scholar] [CrossRef]
- Guillen Diaz-Maroto, N.; Sanz-Pamplona, R.; Berdiel-Acer, M.; Cimas, F.J.; Garcia, E.; Goncalves-Ribeiro, S.; Albert, N.; Garcia-Vicien, G.; Capella, G.; Moreno, V.; et al. Noncanonical TGF-beta Pathway Relieves the Blockade of IL1beta/TGF-beta-Mediated Crosstalk between Tumor and Stroma: TGF-BR1 and TAK1 Inhibition in Colorectal Cancer. Clin. Cancer Res. 2019, 25, 4466–4479. [Google Scholar] [CrossRef]
- Newsted, D.; Banerjee, S.; Watt, K.; Nersesian, S.; Truesdell, P.; Blazer, L.L.; Cardarelli, L.; Adams, J.J.; Sidhu, S.S.; Craig, A.W. Blockade of TGF-beta signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Oncoimmunology 2019, 8, e1539613. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, M.; Dunai, C.; Khuat, L.T.; Aguilar, E.G.; Barao, I.; Murphy, W.J. IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation. Cancers 2020, 12, 3189. https://doi.org/10.3390/cancers12113189
Alvarez M, Dunai C, Khuat LT, Aguilar EG, Barao I, Murphy WJ. IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation. Cancers. 2020; 12(11):3189. https://doi.org/10.3390/cancers12113189
Chicago/Turabian StyleAlvarez, Maite, Cordelia Dunai, Lam T. Khuat, Ethan G. Aguilar, Isabel Barao, and William J. Murphy. 2020. "IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation" Cancers 12, no. 11: 3189. https://doi.org/10.3390/cancers12113189
APA StyleAlvarez, M., Dunai, C., Khuat, L. T., Aguilar, E. G., Barao, I., & Murphy, W. J. (2020). IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation. Cancers, 12(11), 3189. https://doi.org/10.3390/cancers12113189