Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic



Simple Summary
Abstract
1. Introduction
2. Pancreatic Neuroendocrine Tumours (pNET) Cell Lines, Orthotopic Models and Primary Cultures
2.1. pNET Cell Lines
2.2. Orthotopic Models of pNETs
2.3. Primary pNET Cell Cultures
3. Patient-Derived Xenograft (PDX) Models
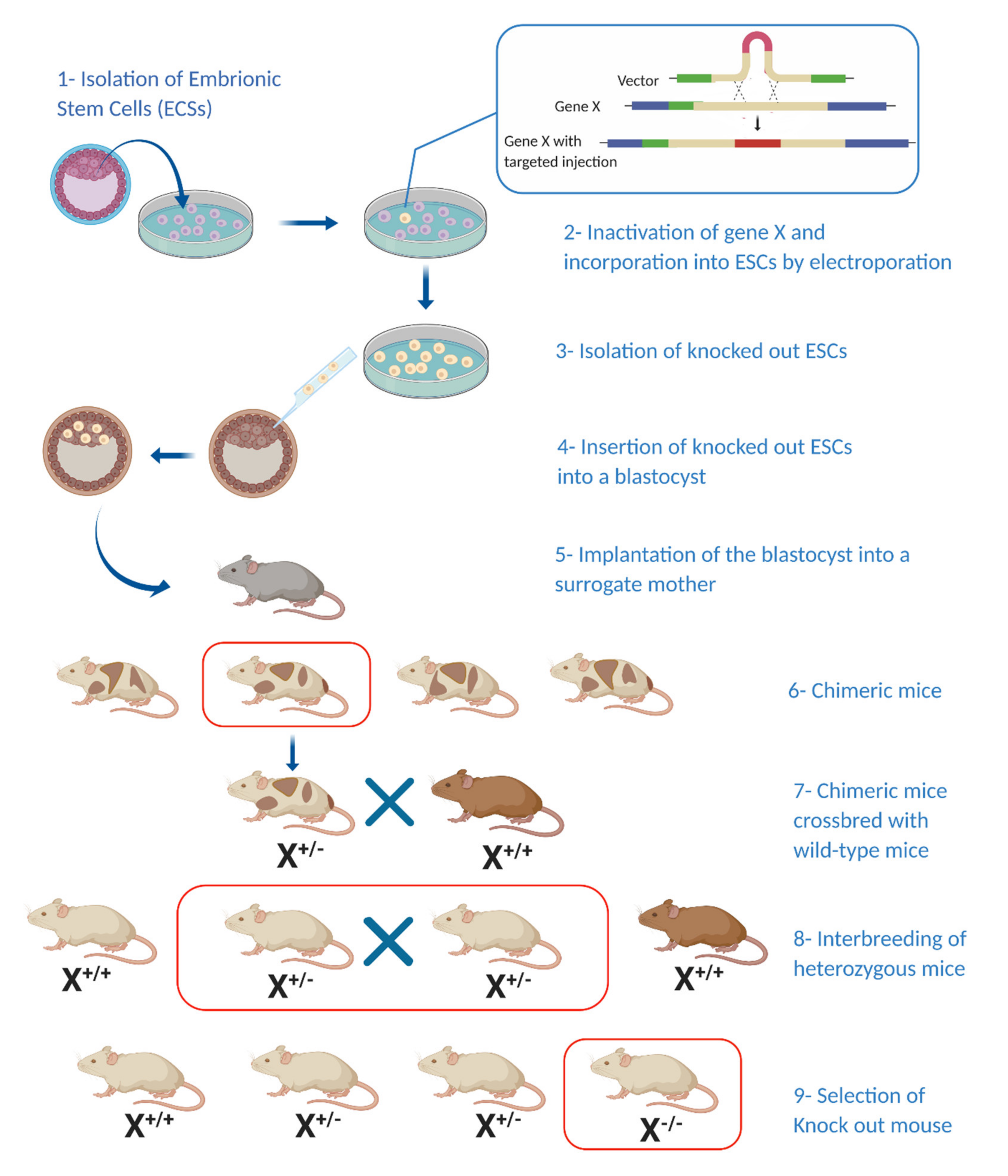
4. Genetically Engineered Mouse Models (GEMMs) of pNETs
4.1. GEMMs of Insulinomas
4.2. GEMMs of Glucagonomas
4.3. GEMMs of Non-Functioning pNETs (NfpNETs)
4.4. GEMMs of Multiple Endocrine Neoplasia 1 (MEN-1)
5. 3D In Vitro Disease Models
5.1. Spheroid-Based 3D Culture Models
5.2. Organoid Culture Models of pNETs
6. The Tumour Microenvironment Is an Important Feature of the Disease Process as a Whole
6.1. Modelling the pNET Microenvironment
6.1.1. The Role of the ECM in pNET Progression
6.1.2. Tissue Martrikines Modulate Disease Processes
6.2. Native Tissue Constructs as Potential Alternatives to Animal Models
Tissue Decellularisation Enables Profiling of the Cancer Matrisome
7. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hopper, A.D.; Jalal, M.; Munir, A. Recent advances in the diagnosis and management of pancreatic neuroendocrine tumours. Front. Gastroenterol. 2018, 10, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Rubin, J.; Farnell, M.B.; Grant, C.S.; Petersen, G.M. Pancreatic endocrine neoplasms: Epidemiology and prognosis of pancreatic endocrine tumors. Endocr. Relat. Cancer 2008, 15, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef] [PubMed]
- Dilley, W.G.; Kalyanaraman, S.; Verma, S.; Cobb, J.P.; Laramie, J.M.; Lairmore, T.C. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol. Cancer 2005, 4, 9. [Google Scholar] [CrossRef]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.; Capdevila, J.A.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.J.; Rindi, G.; Kloppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef]
- Krampitz, G.W.; Norton, J.A. Pancreatic neuroendocrine tumors. Curr. Probl. Surg. 2013, 50, 509–545. [Google Scholar] [CrossRef]
- Turaga, K.K.; Kvols, L.K. Recent progress in the understanding, diagnosis, and treatment of gastroenteropancreatic neuroendocrine tumors. CA A Cancer J. Clin. Am. Cancer Soc. 2011, 61, 113–132. [Google Scholar] [CrossRef]
- Skelin, M.; Rupnik, M.; Cencic, A. Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX 2010, 27, 105–113. [Google Scholar] [CrossRef]
- Townsend, C.M.; Ishizuka, J.; Thompson, J.C. Studies of Growth Regulation in a Neuroendocrine Cell Line. Acta Oncol. 1993, 32, 125–130. [Google Scholar] [CrossRef]
- Kaku, M.; Nishiyama, T.; Yagawa, K.; Abe, M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gan 1980, 71, 596–601. [Google Scholar] [PubMed]
- Zitzmann, K.; De Toni, E.N.; Brand, S.; Göke, B.; Meinecke, J.; Spöttl, G.; Meyer, H.H.; Auernhammer, C.J. The Novel mTOR Inhibitor RAD001 (Everolimus) Induces Antiproliferative Effects in Human Pancreatic Neuroendocrine Tumor Cells. Neuroendocrinology 2007, 85, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.; Beyens, M.; De Beeck, K.O.; Dogan, F.; Van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; De Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Valentino, J.D.; Li, J.; Zaytseva, Y.Y.; Mustain, W.C.; Elliott, V.A.; Kim, J.T.; Harris, J.W.; Campbell, K.; Weiss, H.; Wang, C.; et al. Cotargeting the PI3K and RAS pathways for the treatment of neuroendocrine tumors. Clin. Cancer Res. 2014, 20, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Were, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef]
- Baroni, M.G.; Cavallo, M.G.; Mark, M.; Monetini, L.; Stoehrer, B.; Pozzilli, P. Beta-cell gene expression and functional characterisation of the human insulinoma cell line CM. J. Endocrinol. 1999, 161, 59–68. [Google Scholar] [CrossRef]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef]
- Hofving, T.; Arvidsson, Y.; Almobarak, B.; Inge, L.; Pfragner, R.; Persson, M.; Stenman, G.; Kristiansson, E.; Johanson, V.; Nilsson, O. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr. Relat. Cancer. 2018, 25, X1–X2. [Google Scholar] [CrossRef]
- Siddique, Z.-L.; Drozdov, I.; Floch, J.; Gustafsson, B.I.; Stunes, K.A.; Pfragner, R.; Kidd, M.; Modlin, I.M. KRJ-I and BON Cell Lines: Defining an Appropriate Enterochromaffin Cell Neuroendocrine Tumor Model. Neuroendocrinology 2009, 89, 458–470. [Google Scholar] [CrossRef]
- Luley, K.; Biedermann, S.B.; Künstner, A.; Busch, H.; Franzenburg, S.; Schrader, J.; Grabowski, P.; Wellner, U.F.; Keck, T.; Brabant, G.; et al. A Comprehensive Molecular Characterization of the Pancreatic Neuroendocrine Tumor Cell Lines BON-1 and QGP-1. Cancers 2020, 12, 691. [Google Scholar] [CrossRef]
- Leu, F.P.; Nandi, M.; Niu, C. The effect of transforming growth factor beta on human neuroendocrine tumor BON cell proliferation and differentiation is mediated through somatostatin signaling. Mol. Cancer Res. 2008, 6, 1029–1042. [Google Scholar] [CrossRef]
- Exner, S.; Prasad, V.; Wiedenmann, B.; Groetzinger, C. Octreotide Does Not Inhibit Proliferation in Five Neuroendocrine Tumor Cell Lines. Front. Endocrinol. 2018, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.F.; Radojewski, P.; Marincek, N.; Ben-Shlomo, A.; Grotzky, A.; Olariu, C.I.; Perren, A.; Stettler, C.; Krause, T.M.; Meier, L.P.; et al. Upregulation of Key Molecules for Targeted Imaging and Therapy. J. Nucl. Med. 2016, 57, 1805–1810. [Google Scholar] [CrossRef]
- Yamada, K.M.; Cukierman, E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Hausser, H.-J.; Brenner, R.E. Phenotypic instability of Saos-2 cells in long-term culture. Biochem. Biophys. Res. Commun. 2005, 333, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2015, 14, 3–13. [Google Scholar] [CrossRef]
- Evers, B.M.; Townsend, C.M.; Upp, J.R.; Allen, E.; Hurlbut, S.C.; Kim, S.W.; Rajaraman, S.; Singh, P.; Reubi, J.C.; Thompson, J.C. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology 1991, 101, 303–311. [Google Scholar] [CrossRef]
- Scholz, A.; Wagner, K.; Welzel, M.; Remlinger, F.; Wiedenmann, B.; Siemeister, G.; Rosewicz, S.; Detjen, K.M. The oral multitarget tumour growth inhibitor, ZK 304709, inhibits growth of pancreatic neuroendocrine tumours in an orthotopic mouse model. Gut 2008, 58, 261–270. [Google Scholar] [CrossRef]
- Wu, Y.; Tedesco, L.; Lucia, K.; Schlitter, A.M.; Garcia, J.M.; Esposito, I.; Auernhammer, C.J.; Theodoropoulou, M.; Arzt, E.; Renner, U.; et al. RSUME is implicated in tumorigenesis and metastasis of pancreatic neuroendocrine tumors. Oncotarget 2016, 7, 57878–57893. [Google Scholar] [CrossRef]
- Mohamed, A.; Romano, D.; Saveanu, A.; Roche, C.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Garcia, S.; et al. Anti-proliferative and anti-secretory effects of everolimus on human pancreatic neuroendocrine tumors primary cultures: Is there any benefit from combination with somatostatin analogs? Oncotarget 2017, 8, 41044–41063. [Google Scholar] [CrossRef]
- Mohamed, A.; Blanchard, M.-P.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Monges, G.; Garcia, S.; Ferone, D.; et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in human pancreatic neuroendocrine tumor cultures. Endocr. Relat. Cancer 2014, 21, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Falletta, S.; Partelli, S.; Rubini, C.; Nann, D.; Doria, A.; Marinoni, I.; Polenta, V.; Di Pasquale, C.; Degli Uberti, E.; Perren, A.; et al. mTOR inhibitors response and mTOR pathway in pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 883–891. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; De Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, L.; Serra, S.; Law, C.; Wei, A.; Stockley, T.L.; Ezzat, S.; Asa, S.L. Establishment and Characterization of a Human Neuroendocrine Tumor Xenograft. Endocr. Pathol. 2016, 27, 97–103. [Google Scholar] [CrossRef]
- Dong, X.; Guan, J.; English, J.C.; Flint, J.; Yee, J.; Evans, K.; Murray, N.; MacAulay, C.E.; Ng, R.T.; Gout, P.W.; et al. Patient-Derived First Generation Xenografts of Non-Small Cell Lung Cancers: Promising Tools for Predicting Drug Responses for Personalized Chemotherapy. Clin. Cancer Res. 2010, 16, 1442–1451. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 1–15. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Hermann, P.C.; Heeschen, C. Pancreatic cancer stem cells update and future perspectives. Mol. Oncol. 2010, 4, 431–442. [Google Scholar] [CrossRef]
- Kawasaki, K.; Fujii, M.; Sato, T. Gastroenteropancreatic neuroendocrine neoplasms: Genes, therapies and models. Dis. Model. Mech. 2018, 11, dmm029595. [Google Scholar] [CrossRef]
- Chamberlain, C.E.; German, M.S.; Yang, K.; Wang, J.; VanBrocklin, H.; Regan, M.; Shokat, K.M.; Ducker, G.S.; Kim, G.E.; Hann, B.; et al. A Patient-derived Xenograft Model of Pancreatic Neuroendocrine Tumors Identifies Sapanisertib as a Possible New Treatment for Everolimus-resistant Tumors. Mol. Cancer Ther. 2018, 17, 2702–2709. [Google Scholar] [CrossRef]
- Denayer, T.; Stöhr, T.; Van Roy, M. Animal models in translational medicine: Validation and prediction. Eur. J. Mol. Clin. Med. 2014, 2, 5. [Google Scholar] [CrossRef]
- Lines, K.E.; Nunes, R.P.V.; Frost, M.; Yates, C.J.; Stevenson, M.; Thakker, R.V. A MEN1 pancreatic neuroendocrine tumour mouse model under temporal control. Endocr. Connect. 2017, 6, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nat. Cell Biol. 1985, 315, 115–122. [Google Scholar] [CrossRef]
- Onrust, S.V.; Hartl, P.M.; Rosen, S.D.; Hanahan, D. Modulation of L-selectin ligand expression during an immune response accompanying tumorigenesis in transgenic mice. J. Clin. Investig. 1996, 97, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Contractor, T.; Vosburgh, E.; Du, Y.-C.N.; Tang, L.H.; Clausen, R.; Harris, C.R. Alleles of Insm1 determine whether RIP1-Tag2 mice produce insulinomas or nonfunctioning pancreatic neuroendocrine tumors. Oncology 2019, 8, 16. [Google Scholar] [CrossRef]
- McHugh, K.E.; Mukhopadhyay, S.; Doxtader, E.E.; Lanigan, C.; Allende, D.S. INSM1 Is a Highly Specific Marker of Neuroendocrine Differentiation in Primary Neoplasms of the Gastrointestinal Tract, Appendix, and Pancreas. Am. J. Clin. Pathol. 2020, 153, 811–820. [Google Scholar] [CrossRef]
- Staaf, J.; Tran, L.; Söderlund, L.; Nodin, B.; Jirström, K.; Vidarsdottir, H.; Planck, M.; Mattsson, J.S.M.; Botling, J.; Micke, P.; et al. Diagnostic Value of Insulinoma-Associated Protein 1 (INSM1) and Comparison With Established Neuroendocrine Markers in Pulmonary Cancers: A Comprehensive Study and Review of the Literature. Arch. Pathol. Lab. Med. 2020, 1, 1075–1085. [Google Scholar] [CrossRef]
- Tanigawa, M.; Nakayama, M.; Taira, T.; Hattori, S.; Mihara, Y.; Kondo, R.; Kusano, H.; Nakamura, K.; Abe, Y.; Ishida, Y.; et al. Insulinoma-associated protein 1 (INSM1) is a useful marker for pancreatic neuroendocrine tumor. Med. Mol. Morphol. 2017, 51, 32–40. [Google Scholar] [CrossRef]
- Alliouachene, S.; Tuttle, R.L.; Boumard, S.; Lapointe, T.; Berissi, S.; Germain, S.; Jaubert, F.; Tosh, D.; Birnbaum, M.J.; Pende, M. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J. Clin. Investig. 2008, 118, 3629–3638. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-Induced Apoptosis in β Cells Exposes Multiple Oncogenic Properties of Myc and Triggers Carcinogenic Progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef]
- Brubaker, P.L.; Lee, Y.C.; Drucker, D.J. Alterations in proglucagon processing and inhibition of proglucagon gene expression in transgenic mice which contain a chimeric proglucagon-SV40 T antigen gene. J. Biol. Chem. 1992, 267, 20728–20733. [Google Scholar] [PubMed]
- Rindi, G.; Efrat, S.; Ghatei, M.A.; Bloom, S.R.; Solcia, E.; Polak, J.M. Glucagonomas of transgenic mice express a wide range of general neuroendocrine markers and bioactive peptides. Virchows Arch. 1991, 419, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Glenn, S.T.; Jones, C.A.; Sexton, S.; LeVea, C.M.; Caraker, S.M.; Hajduczok, G.; Gross, K.W. Conditional deletion of p53 and Rb in the renin-expressing compartment of the pancreas leads to a highly penetrant metastatic pancreatic neuroendocrine carcinoma. Oncogene 2013, 33, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Kasai, K.; Takagishi, Y.; Kikumori, T.; Imai, T.; Murata, Y.; Hayashi, Y. Pancreatic Neuroendocrine Tumors in Mice Deficient in Proglucagon-Derived Peptides. PLoS ONE 2015, 10, e0133812. [Google Scholar] [CrossRef] [PubMed]
- Saloustros, E.; Salpea, P.; Starost, M.; Liu, S.; Faucz, F.R.; London, E.; Szarek, E.; Song, W.-J.; Hussain, M.; Stratakis, C.A. Prkar1a gene knockout in the pancreas leads to neuroendocrine tumorigenesis. Endocr. Relat. Cancer 2017, 24, 31–40. [Google Scholar] [CrossRef]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and Abnormal Functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef]
- Biondi, C.A.; Gartside, M.G.; Waring, P.; Loffler, K.A.; Stark, M.S.; Magnuson, M.A.; Kay, G.F.; Hayward, N.K. Conditional Inactivation of the Men1 Gene Leads to Pancreatic and Pituitary Tumorigenesis but Does Not Affect Normal Development of These Tissues. Mol. Cell. Biol. 2004, 24, 3125–3131. [Google Scholar] [CrossRef]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2019, 27, 269–283. [Google Scholar] [CrossRef]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J. Cell Sci. 2016, 130, 203–218. [Google Scholar] [CrossRef]
- Nyga, A.; Cheema, U.; Loizidou, M. 3D tumour models: Novel in vitro approaches to cancer studies. J. Cell Commun. Signal. 2011, 5, 239–248. [Google Scholar] [CrossRef]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas2. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, M.H.; Lee, J.H.; Seliktar, D.; Cho, N.-J.; Tan, L.P. Modulation of Huh7.5 Spheroid Formation and Functionality Using Modified PEG-Based Hydrogels of Different Stiffness. PLoS ONE 2015, 10, e0118123. [Google Scholar] [CrossRef]
- Lin, R.-Z.; Chou, L.-F.; Chien, C.-C.M.; Chang, H.-Y. Dynamic analysis of hepatoma spheroid formation: Roles of E-cadherin and β1-integrin. Cell Tissue Res. 2006, 324, 411–422. [Google Scholar] [CrossRef]
- Hamilton, G. Multicellular spheroids as an in vitro tumor model. Cancer Lett. 1998, 131, 29–34. [Google Scholar] [CrossRef]
- Torisawa, Y.-S.; Takagi, A.; Shiku, H.; Yasukawa, T.; Matsue, T. A multicellular spheroid-based drug sensitivity test by scanning electrochemical microscopy. Oncol. Rep. 2005, 13, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Wen, Z.; Liao, Q.; Hu, Y.; You, L.; Zhou, L.; Zhao, Y. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Braz. J. Med. Biol. Res. 2013, 46, 634–642. [Google Scholar] [CrossRef]
- Lazzari, G.; Nicolas, V.; Matsusaki, M.; Akashi, M.; Couvreur, P.; Mura, S. Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 2018, 78, 296–307. [Google Scholar] [CrossRef]
- Bresciani, G.; Hofland, L.J.; Dogan, F.; Giamas, G.; Gagliano, T.; Zatelli, M.C. Evaluation of Spheroid 3D Culture Methods to Study a Pancreatic Neuroendocrine Neoplasm Cell Line. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Cortez, E.; Gladh, H.; Braun, S.; Bocci, M.; Cordero, E.; Björkström, N.K.; Miyazaki, H.; Michael, I.P.; Eriksson, U.; Folestad, E.; et al. Functional malignant cell heterogeneity in pancreatic neuroendocrine tumors revealed by targeting of PDGF-DD. Proc. Natl. Acad. Sci. USA 2016, 113, E864–E873. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Martinez, A.; Van, D.; Dogan, F.; Van, K.P.; Castanño, J.; Feelders, R.; Hofland, L. Utility of a 3D spheroid cell culture system in neuroendocrine tumors. Endocr. Abstr. 2018. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2014, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Benton, G.; Arnaoutova, I.; George, J.; Kleinman, H.K.; Koblinski, J. Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 2014, 79, 3–18. [Google Scholar] [CrossRef]
- Benton, G.J.; Kleinman, H.K.; George, J.; Arnaoutova, I. Multiple uses of basement membrane-like matrix (BME/Matrigel) in vitro and in vivo with cancer cells. Int. J. Cancer 2011, 128, 1751–1757. [Google Scholar] [CrossRef]
- Li, L. Optimizing a 3D Culture System to Study the Interaction between Epithelial Breast Cancer and Its Surrounding Fibroblasts. J. Cancer 2011, 2, 458. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- April-Monn, S.L.; Wiedmer, T.; Skowronska, M.S.; Maire, R.S.; Lena, M.S.; Trippel, M.; Di Domenico, A.; Muffatti, F.; Andreasi, V.; Capurso, G.; et al. 3D Primary Cell Culture: A Novel Preclinical Model For Pancreatic Neuroendocrine Tumors (PanNETs). Neuroendocrinology 2020. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling Pancreatic Cancer with Organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Colella, G.; Fazioli, F.; Gallo, M.; De Chiara, A.; Apice, G.; Ruosi, C.; Cimmino, A.; De Nigris, F. Sarcoma Spheroids and Organoids—Promising Tools in the Era of Personalized Medicine. Int. J. Mol. Sci. 2018, 19, 615. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Thakuri, P.S.; Liu, C.; Luker, G.D.; Tavana, H. Biomaterials-Based Approaches to Tumor Spheroid and Organoid Modeling. Adv. Healthc. Mater. 2017, 7, e1700980. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2016, 216, 31–40. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Nanki, K.; Sato, T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 2015, 10, 1474–1485. [Google Scholar] [CrossRef]
- Young, M.; Reed, K.R. Organoids as a Model for Colorectal Cancer. Curr. Color. Cancer Rep. 2016, 12, 281–287. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Pelle’, E.; Quaresmini, D.; Rizzo, F.M.; Tucci, M.; Silvestris, F. The Tumor Microenvironment in Neuroendocrine Tumors: Biology and Therapeutic Implications. Neuroendocrinology 2019, 109, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Corti, A. Chromogranin A and the Tumor Microenvironment. Cell. Mol. Neurobiol. 2010, 30, 1163–1170. [Google Scholar] [CrossRef]
- Saupe, F.; Schwenzer, A.; Jia, Y.; Gasser, I.; Spenlé, C.; Langlois, B.; Kammerer, M.; Lefebvre, O.; Hlushchuk, R.; Rupp, T.; et al. Tenascin-C Downregulates Wnt Inhibitor Dickkopf-1, Promoting Tumorigenesis in a Neuroendocrine Tumor Model. Cell Rep. 2013, 5, 482–492. [Google Scholar] [CrossRef]
- Hunter, K.E.; Palermo, C.; Kester, J.C.; Simpson, K.; Li, J.-P.; Tang, L.H.; Klimstra, D.S.; Vlodavsky, I.; Joyce, J.A. Heparanase promotes lymphangiogenesis and tumor invasion in pancreatic neuroendocrine tumors. Oncogene 2013, 33, 1799–1808. [Google Scholar] [CrossRef]
- Maquart, F.-X.; Brassart-Pasco, S.; Ramont, L.; Hornebeck, W.; Monboisse, J.-C. An introduction to matrikines: Extracellular matrix-derived peptides which regulate cell activity. Crit. Rev. Oncol. 2004, 49, 199–202. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Roy, R.; Zurakowski, D.; Wischhusen, J.; Frauenhoffer, C.; Hooshmand, S.; Kulke, M.; Moses, M.A. Urinary TIMP-1 and MMP-2 levels detect the presence of pancreatic malignancies. Br. J. Cancer 2014, 111, 1772–1779. [Google Scholar] [CrossRef]
- Astrof, S.; Crowley, D.; George, E.L.; Fukuda, T.; Sekiguchi, K.; Hanahan, D.; Hynes, R.O. Direct Test of Potential Roles of EIIIA and EIIIB Alternatively Spliced Segments of Fibronectin in Physiological and Tumor Angiogenesis. Mol. Cell. Biol. 2004, 24, 8662–8670. [Google Scholar] [CrossRef]
- Murphy, P.A.; Begum, S.; Hynes, R.O. Tumor Angiogenesis in the Absence of Fibronectin or Its Cognate Integrin Receptors. PLoS ONE 2015, 10, e0120872. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Duncan, M.B.; Pahler, J.; Sugimoto, H.; Martino, M.; Lively, J.; Mundel, T.; Soubasakos, M.; Rubin, K.; Takeda, T.; et al. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc. Natl. Acad. Sci. USA 2011, 108, 9939–9944. [Google Scholar] [CrossRef] [PubMed]
- Parilla, J.H.; Hull, R.L.; Zraika, S. Neprilysin Deficiency Is Associated with Expansion of Islet β-Cell Mass in High Fat-Fed Mice. J. Histochem. Cytochem. 2018, 66, 523–530. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, L.; Sun, Y.; Jiang, Z.; Domsic, J.; An, C.; Xing, B.; Tian, J.; Liu, X.; Metz, D.C.; et al. Menin and Daxx Interact to Suppress Neuroendocrine Tumors through Epigenetic Control of the Membrane Metallo-Endopeptidase. Cancer Res. 2016, 77, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; MacLeod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of treatment effects between animal experiments and clinical trials: Systematic review. BMJ 2006, 334, 197. [Google Scholar] [CrossRef]
- Ford, K.A. Refinement, Reduction, and Replacement of Animal Toxicity Tests by Computational Methods. ILAR J. 2016, 57, 226–233. [Google Scholar] [CrossRef]
- Mazza, G.; Rombouts, K.; Hall, A.R.; Urbani, L.; Luong, T.V.; Al-Akkad, W.; Longato, L.; Brown, D.; Maghsoudlou, P.; Dhillon, A.P.; et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci. Rep. 2015, 5, 13079. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Ott, H.C. Organ engineering based on decellularized matrix scaffolds. Trends Mol. Med. 2011, 17, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef]
- Pearce, O.M.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2017, 8, 304–319. [Google Scholar] [CrossRef]
- Naba, A.; Pearce, O.M.T.; Del Rosario, A.; Ma, D.; Ding, H.; Rajeeve, V.; Cutillas, P.R.; Balkwill, F.; Hynes, R.O. Characterization of the Extracellular Matrix of Normal and Diseased Tissues Using Proteomics. J. Proteome Res. 2017, 16, 3083–3091. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Mani, D.R.; Carr, S.A.; Hynes, R.O. Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci. Rep. 2017, 7, 40495. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, A.M.; Amenta, P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995, 9, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Hinds, S.; Bian, W.; Dennis, R.G.; Bursac, N. The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 2011, 32, 3575–3583. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.M.; Hayda, K.N.; Anseth, K.S. Cell-matrix interactions improve beta-cell survival and insulin secretion in three-dimensional culture. Tissue Eng. Part A 2008, 14, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Guyette, J.P.; Gilpin, S.E.; Charest, J.M.; Tapias, L.F.; Ren, X.; Ott, H.C. Perfusion decellularization of whole organs. Nat. Protoc. 2014, 9, 1451–1468. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, R.G.; Vermette, P. Decellularized pancreas as a native extracellular matrix scaffold for pancreatic islet seeding and culture. J. Tissue Eng. Regen. Med. 2018, 12, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.-K.; Bertera, S.; Olsen, P.; Candiello, J.E.; Halfter, W.; Uechi, G.; Balasubramani, M.; Johnson, S.A.; Sicari, B.M.; Kollar, E.; et al. Perfusion-decellularized pancreas as a natural 3D scaffold for pancreatic tissue and whole organ engineering. Biomaterials 2013, 34, 6760–6772. [Google Scholar] [CrossRef]
- Peloso, A.; Urbani, L.; Cravedi, P.; Katari, R.; Maghsoudlou, P.; Fallas, M.E.A.; Sordi, V.; Citro, A.; Purroy, C.; Niu, G.; et al. The Human Pancreas as a Source of Protolerogenic Extracellular Matrix Scaffold for a New-generation Bioartificial Endocrine Pancreas. Ann. Surg. 2016, 264, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Ye, K.; Jin, S. Proteomic analysis of decellularized pancreatic matrix identifies collagen V as a critical regulator for islet organogenesis from human pluripotent stem cells. Biomaterials 2020, 233, 119673. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.H.; Park, K.M.; Ghim, J.H.; Yang, S.R.; Woo, H.M. Three dimensional culture of HepG2 liver cells on a rat decellularized liver matrix for pharmacological studies. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 104, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Werner, M.E.; Roche, K.C.; Hanson, A.D.; Foote, H.P.; Yu, S.K.; Warner, S.; Copp, J.A.; Lara, H.; Wauthier, E.L.; et al. Organ-specific metastases obtained by culturing colorectal cancer cells on tissue-specific decellularized scaffolds. Nat. Biomed. Eng. 2018, 2, 443–452. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.-G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Liu, G.; Wang, B.; Li, S.; Jin, Q.; Dai, Y. Human breast cancer decellularized scaffolds promote epithelial-to-mesenchymal transitions and stemness of breast cancer cells in vitro. J. Cell. Physiol. 2018, 234, 9447–9456. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cells of Origin | Biological Properties | Advantages and Potential Use | Disadvantages | |
|---|---|---|---|---|
| Spheroids | Cell lines [83,84,85] PDX cells [86] | Cultured in suspension in basement membrane elements Spheroid size and compactness vary between cell lines [85] Mimic cell–cell interactions that promote proliferation and survival observed in vivo [86] Trilaminar structure (Proliferative, quiescent, and necrotic zones) [85] Dynamic growth rate similar to those observed in vivo: early exponential phase with cell proliferation followed by a decline in growth with increased necrosis [85] | Recapitulates mismatch between uncontrolled growth and nutrient supply [85] Hypoxic environment in spheroid core promotes evolution of cancer stem cells [86] The use of co-cultures with stromal components enables studying signalling pathways, angiogenesis and invasion [85] Ideal for high-throughput drug screening in more in vivo like spatial arrangement of cells compared to monolayer cultures [86] | Limited clinical value due to the absence of biological complexity and functionality of in vivo tumours [83] |
| Organoids | Cells derived from dissociated primary tissue [83,86] Stem cells [87] Genetic manipulation of cells derived from normal tissues [88,89] | Cultured in suspension in basement membrane elements Self-organising or organogenesis-cue-dependent Reproduce functionality and architecture of the tissue of origin [86] Preservation of genetic expression and histopathologic features of the original tumour [86] | Offer a tumour-specific and personalised approach to treatment [86] Orthotopic transplantation of organoids in mice reproduces primary tumour properties [36] Creation of organoid biobanks of tumours derived from patients with different tumour phenotypes [36] Study the role of infectious pathogens in tumourigenesis [36] Modelling of cancer associated genetic aberrations using CRISPR/Cas9 genome editing [88] Study the effect of drugs in a way more similar to the conditions in vivo compared to 2D models [86] | Risk of genetic drift from original tumour due to instability of tumour genome. This process can be reduced by the restriction of culture time [90] Genetically engineered organoids show lower metastatic potential compared to those derived from primary tumours [88] Lack vasculature and immune components [36,86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ney, A.; Canciani, G.; Hsuan, J.J.; Pereira, S.P. Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic. Cancers 2020, 12, 3170. https://doi.org/10.3390/cancers12113170
Ney A, Canciani G, Hsuan JJ, Pereira SP. Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic. Cancers. 2020; 12(11):3170. https://doi.org/10.3390/cancers12113170
Chicago/Turabian StyleNey, Alexander, Gabriele Canciani, J. Justin Hsuan, and Stephen P. Pereira. 2020. "Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic" Cancers 12, no. 11: 3170. https://doi.org/10.3390/cancers12113170
APA StyleNey, A., Canciani, G., Hsuan, J. J., & Pereira, S. P. (2020). Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic. Cancers, 12(11), 3170. https://doi.org/10.3390/cancers12113170