Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein

 ,
, 
Simple Summary
Abstract
1. Introduction
2. Results
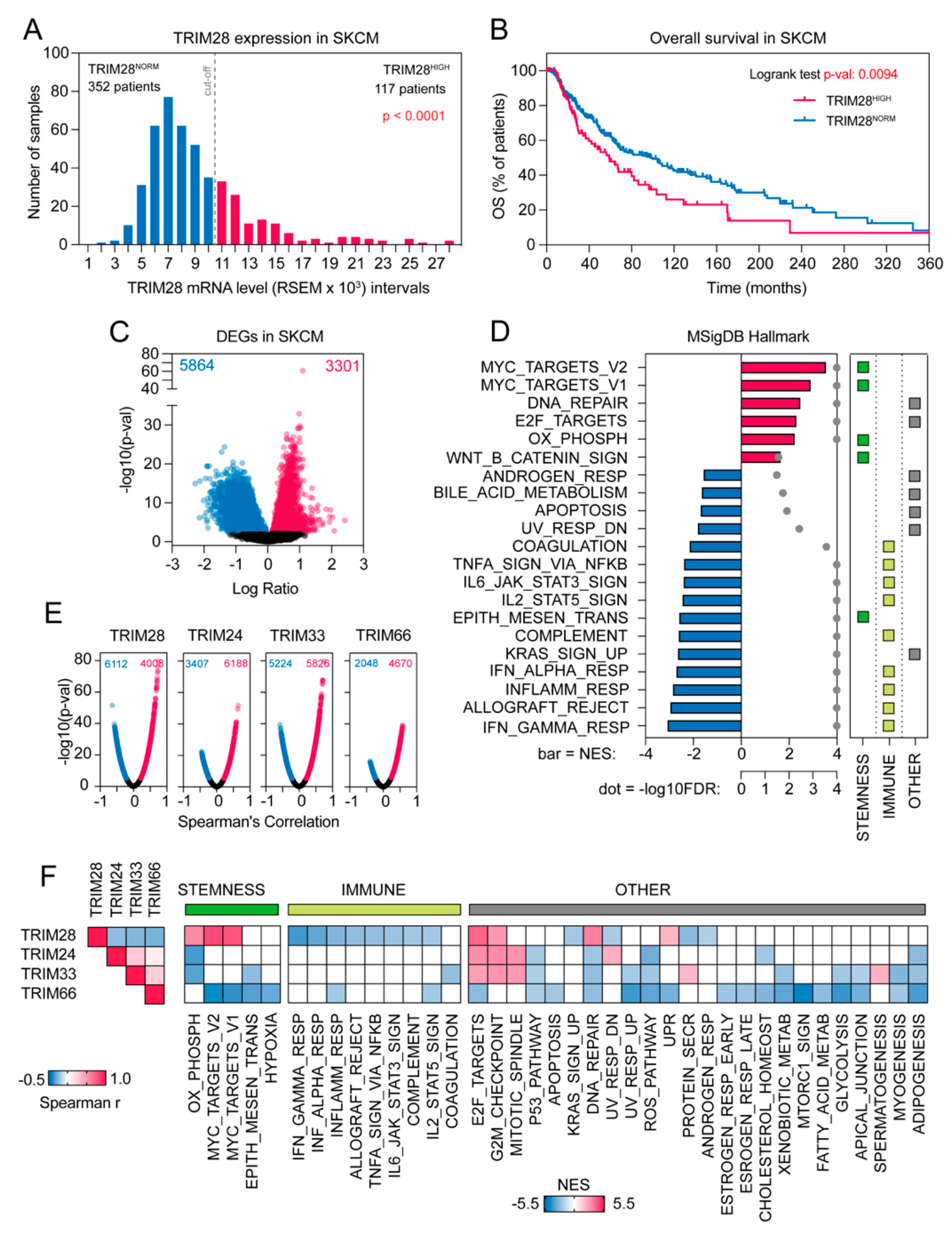
2.1. The Transcriptome Profile of TRIM28 High Expressing Melanoma Patients Negatively Correlates with Immune-Associated Gene Signatures While Being Significantly Enriched with Stemness-Associated Biological Processes
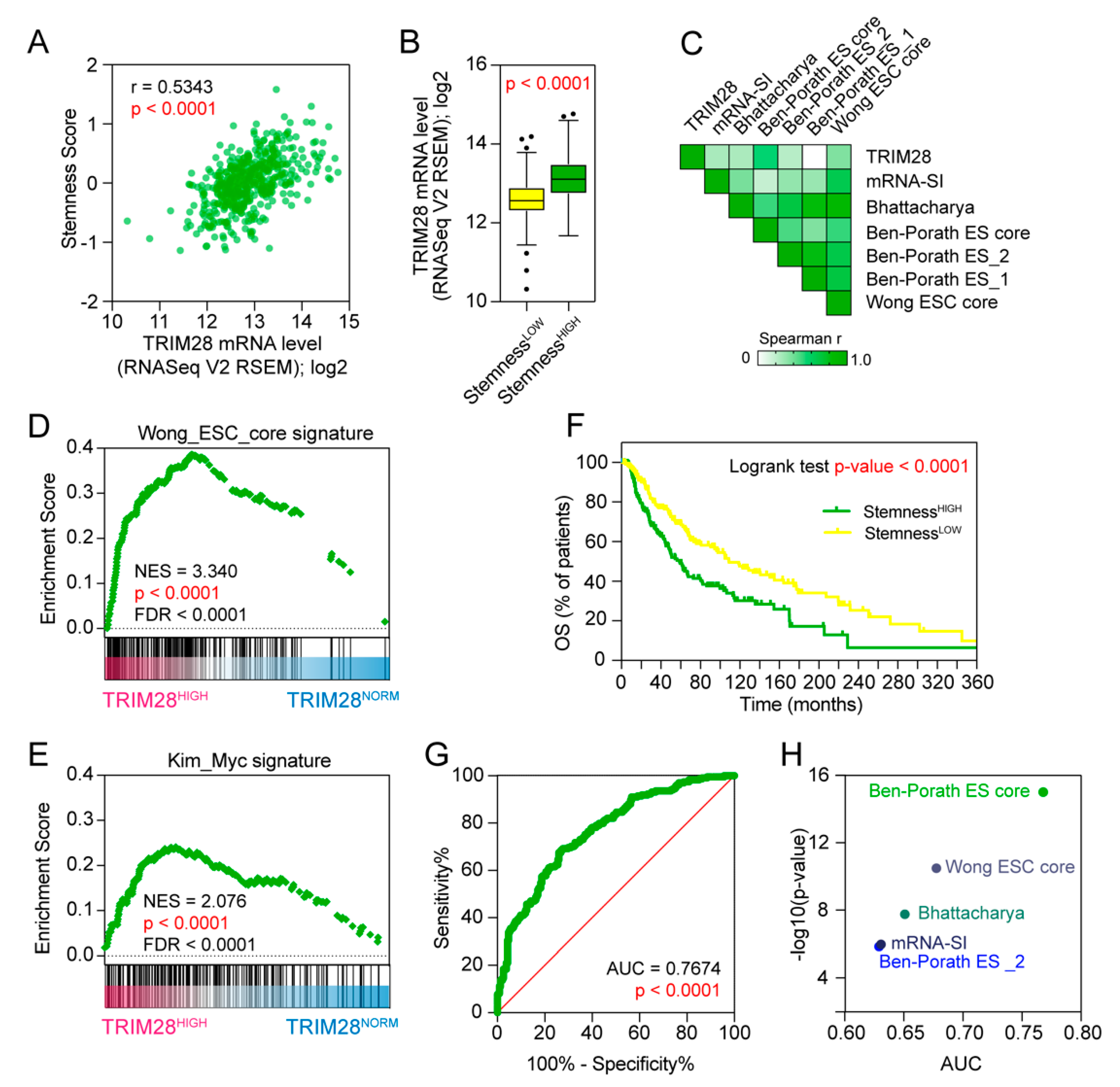
2.2. TRIM28 High Expressing Melanomas Are Dedifferentiated Tumors Enriched with Stem Cell-Associated Features
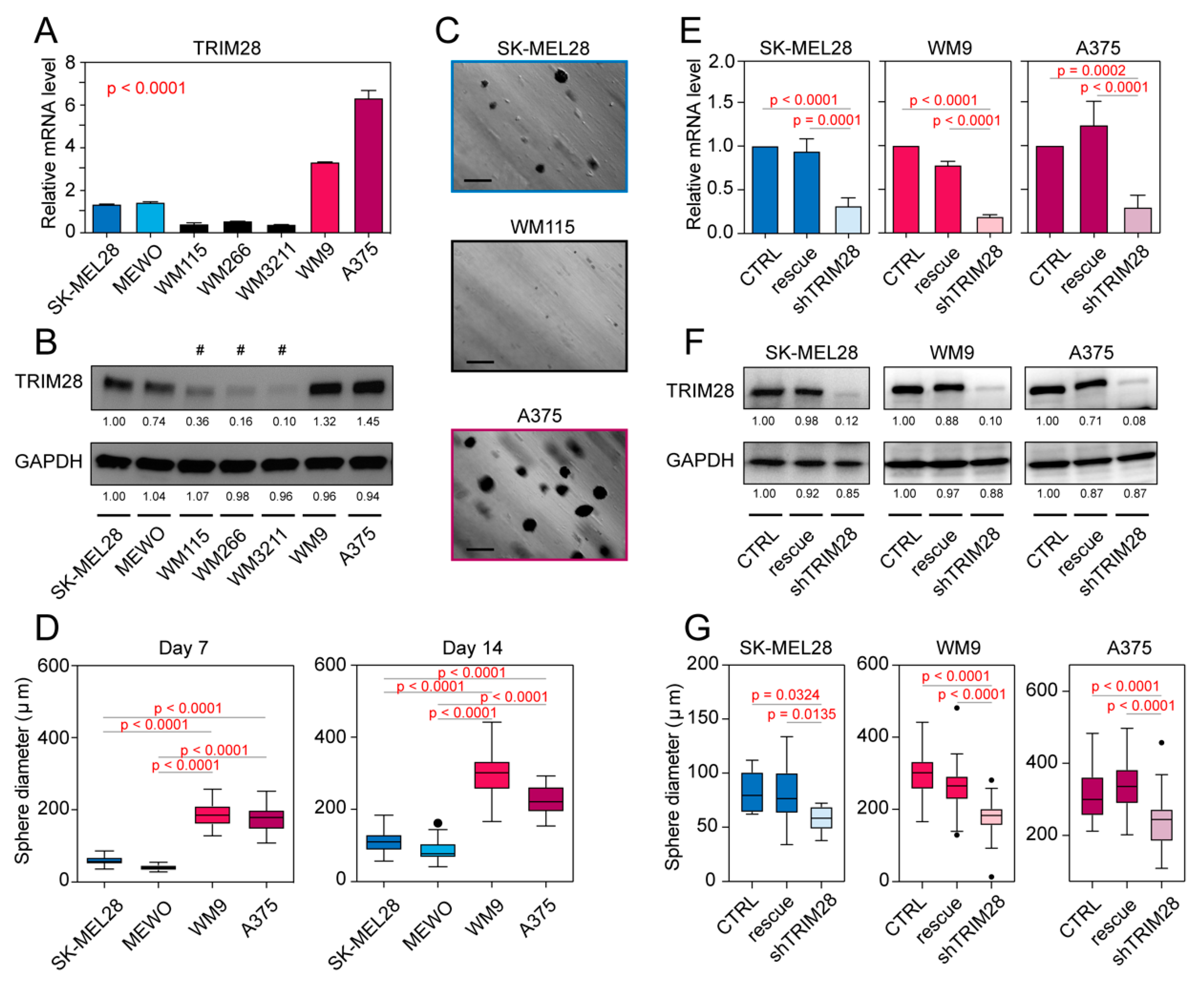
2.3. TRIM28 High Expressing Melanoma Cell Lines Possess a Higher Potential of Melanosphere Formation
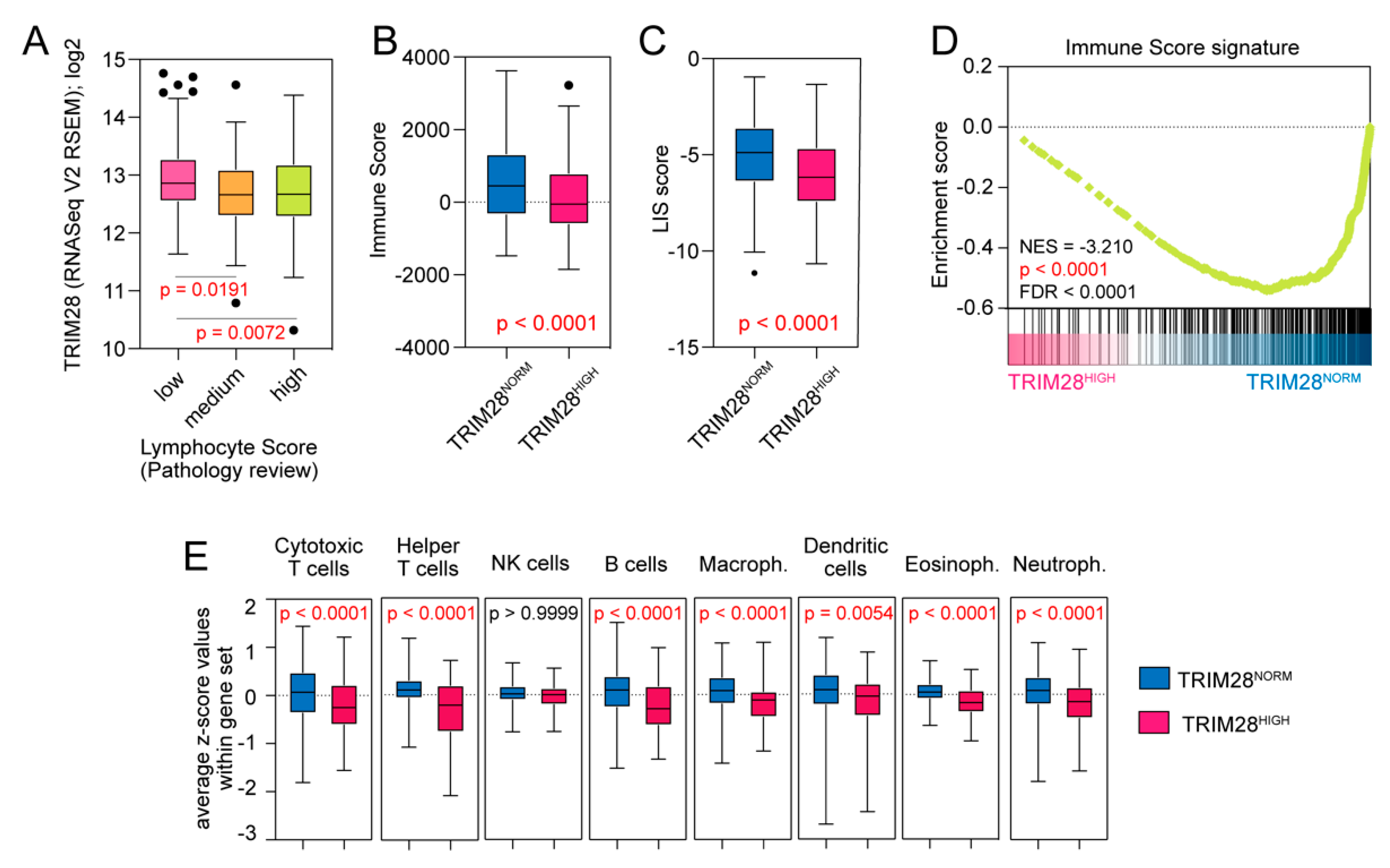
2.4. TRIM28 High Expressing Melanomas Are Significantly Depleted with Tumor-Infiltrating Lymphocytes
2.5. TRIM28 Expression Is Strictly Associated with Stemness-High/Immune-Low Melanoma Phenotype and Can Predict the Survival of Melanoma Patients
2.6. Significant Attenuation of the Interferon Signaling by TRIM28-Mediated Epigenetic Silencing of the IRF Transcription Factor Family Might Facilitate Stemness High/Immune Low Melanoma Phenotype
3. Discussion
4. Materials and Methods
4.1. SKCM TCGA Genomic and Clinical Data
4.2. Transcriptomic Data
4.3. Gene Set Enrichment Analysis
4.4. Methylation Data
4.5. Stemness-Associated Scores
4.6. Immune-Associated Scores
4.7. Validation Sets
4.8. Statistical Analyses
4.9. Melanoma Cell Lines
4.10. RT-qPCR Analyses
4.11. Western Blot
4.12. Cell Proliferation Assay
4.13. Sphere Formation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Begg, G.E.; Schultz, D.C.; Friedman, J.R.; Jensen, D.E.; Speicher, D.W.; Rauscher, F.J., III. Reconstitution of the KRAB-KAP-1 repressor complex: A model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 2000, 295, 1139–1162. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, 25361–25369. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.X.; El Farran, C.A.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, Y.F.; Goh, G.Y.L.; Neo, S.P.; et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef]
- Venkov, C.D.; Link, A.J.; Jennings, J.L.; Plieth, D.; Inoue, T.; Nagai, K.; Xu, C.; Dimitrova, Y.N.; Rauscher, F.J., III; Neilson, E.G. A proximal activator of transcription in epithelial-mesenchymal transition. J. Clin. Investig. 2007, 117, 482–491. [Google Scholar] [CrossRef]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J., III; Chen, J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.-G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef]
- Klimczak, M.; Czerwińska, P.; Mazurek, S.; Sozańska, B.; Biecek, P.; Mackiewicz, A.; Wiznerowicz, M. TRIM28 epigenetic corepressor is indispensable for stable induced pluripotent stem cell formation. Stem Cell Res. 2017, 23, 163–172. [Google Scholar] [CrossRef]
- Oleksiewicz, U.; Gładych, M.; Raman, A.T.; Heyn, H.; Mereu, E.; Chlebanowska, P.; Andrzejewska, A.; Sozańska, B.; Samant, N.; Fąk, K.; et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Rep. 2017, 9, 2065–2080. [Google Scholar] [CrossRef]
- Czerwińska, P.; Kamińska, B. Regulation of breast cancer stem cell features. Contemp. Oncol. 2015, 19, A7–A15. [Google Scholar] [CrossRef]
- Lee, G.; Hall, R.R., III; Ahmed, A.U. Cancer Stem Cells: Cellular Plasticity, Niche, and its Clinical Relevance. J. Stem. Cell Res. Ther. 2016, 6, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. Application of induced pluripotency in cancer studies. Rep. Pract. Oncol. Radiother. 2018, 23, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Addison, J.B.; Koontz, C.; Fugett, J.H.; Creighton, C.J.; Chen, D.; Farrugia, M.K.; Padon, R.R.; Voronkova, M.A.; McLaughlin, S.L.; Livengood, R.H.; et al. KAP1 Promotes Proliferation and Metastatic Progression of Breast Cancer Cells. Cancer Res. 2014, 75, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozańska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008, 2, 333–344. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Miura, T.; Brandenberger, R.; Mejido, J.; Luo, Y.; Yang, A.X.; Joshi, B.H.; Ginis, I.; Thies, R.S.; Amit, M.; et al. Gene expression in human embryonic stem cell lines: Unique molecular signature. Blood 2004, 103, 2956–2964. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Becht, E.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.A.; Czerwinska, P. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem Cells 2020, 38, 165–173. [Google Scholar]
- Cirenajwis, H.; Ekedahl, H.; Lauss, M.; Harbst, K.; Carneiro, A.; Enoksson, J.; Rosengren, F.; Linda Werner-Hartman, L.; Törngren, T.; Kvist, A.; et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 2015, 6, 12297–12309. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.-L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Yu, C.-C.; Wang, B.-Y.; Chang, W.-W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli., R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.; Plaisier, C.L.; Eddy, J.A.; et al. Resource The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schaafsma, E.; Gorlov, I.P.; Hernando, E.; Thomas, N.E.; Shen, R.; Turk, M.J.; Berwick, M.; Amos, C.I.; Cheng, C. A Leukocyte Infiltration Score Defined by a Gene Signature Predicts Melanoma Patient Prognosis. Mol. Cancer Res. 2019, 17, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Kamitani, S.; Ohbayashi, N.; Ikeda, O.; Togi, S.; Muromoto, R.; Sekine, Y.; Ohta, K.; Ishiyama, H.; Matsuda, T. Biochemical and Biophysical Research Communications KAP1 regulates type I interferon / STAT1-mediated IRF-1 gene expression. Biochem. Biophys. Res. Commun. 2008, 370, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Halada, P.; Hernychová, L.; Chong, Y.P.; Žáková, J.; Hupp, T.R.; Vojtesek, B.; Ball, K.L. A multiprotein binding interface in an intrinsically disordered region of the tumor suppressor protein interferon regulatory factor-1. J. Biol. Chem. 2011, 286, 14291–14303. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermot, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, S.; Fu, X.; Feng, J.; Xu, S.; Ying, G. High Levels of KAP1 Expression Are Associated with Aggressive Clinical Features in Ovarian Cancer. Int. J. Mol. Sci. 2015, 16, 363–377. [Google Scholar] [CrossRef]
- Hu, M.; Fu, X.; Cui, Y.; Xu, S.; Xu, Y.; Dong, Q.; Sun, L. Expression of KAP1 in epithelial ovarian cancer and its correlation with drug-resistance. Int. J. Clin. Exp. Med. 2015, 8, 17308–17320. [Google Scholar]
- Wang, Y.; Jiang, J.; Li, Q.; Ma, H.; Xu, Z.; Gao, Y. KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int. J. Clin. Oncol. 2016, 21, 927–933. [Google Scholar] [CrossRef]
- Fernandez-Marrero, Y.; Bachmann, D.; Lauber, E.; Kaufmann, T. Negative Regulation of BOK Expression by Recruitment of TRIM28 to Regulatory Elements in Its 3′ Untranslated Region. IScience 2018, 9, 461–474. [Google Scholar] [CrossRef]
- Lee, A.K.; Pan, D.; Bao, X.; Hu, M.; Li, F.; Li, C.-Y. Endogenous Retrovirus Activation as a Key Mechanism of Anti-Tumor Immune Response in Radiotherapy. Radiat. Res. 2020, 193, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Kurisaki, A.; Watanabe-Susaki, K.; Nakajima, Y.; Nakanishi, M.; Arai, Y.; Shiota, K.; Sugino, H.; Asashima, M. TIF1beta regulates the pluripotency of embryonic stem cells in a phosphorylation-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 10926–10931. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Ren, X.; Kerppola, T.K. KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Mol. Cell Biol. 2014, 34, 2075–2091. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Kim, J.; Xu, Q.; Leng, Y.; Orkin, S.H.; Elledge, S.J. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009, 23, 837–848. [Google Scholar] [CrossRef]
- Wiznerowicz, M.; Jakobsson, J.; Szulc, J.; Liao, S.; Quazzola, A.; Beermann, F.; Aebischer, P.; Trono, D. The Krüppel-associated box repressor domain can trigger de novo promoter methylation during mouse early embryogenesis. J. Biol. Chem. 2007, 282, 34535–34541. [Google Scholar] [CrossRef]
- Li, J.; Xi, Y.; Li, W.; McCarthy, R.L.; Stratton, S.A.; Zou, W.; Li, W.; Dent, S.Y.; Jain, A.K.; Barton, M.C. TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene 2017, 36, 2991–3001. [Google Scholar] [CrossRef]
- Wang, C.; Rauscher, F.J., III; Cress, W.D.; Chen, J. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 2007, 282, 29902–29909. [Google Scholar] [CrossRef]
- Chen, L.; Chen, D.T.; Chen, T.; Kurtyka, C.; Rawal, B.; Fulp, W.J.; Haura, E.B.; Cress, W.D. Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J. Biol. Chem. 2012, 287, 40106–40118. [Google Scholar] [CrossRef]
- Pineda, C.T.; Potts, P.R. Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy 2015, 11, 844–846. [Google Scholar] [CrossRef]
- Alavi, S.; Stewart, A.J.; Kefford, R.F.; Lim, S.Y.; Shklovskaya, E.; Rizos, H. Interferon Signaling Is Frequently Downregulated in Melanoma. Front. Immunol. 2018, 9, 1414. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- GSEA. Available online: http://www.broad.mit.edu/gsea/index.html (accessed on 10 April 2020).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- MSigDB. Available online: http://www.broad.mit.edu/gsea/.msigdb/msigdb_index.html (accessed on 10 April 2020).
- Available online: http://mexpress.be (accessed on 20 May 2020).
- Koch, A.; De Meyer, T.; Jeschke, J.; Van Criekinge, W. MEXPRESS: Visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 2015, 16, 636–642. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Feature | TRIM28NORM | TRIM28HIGH | p-Value 1 |
|---|---|---|---|
| Sex: male, n (%) | 222 (0.64) | 65 (0.56) | 0.150 |
| Primary melanoma, n (%) | 74 (21.3) | 34 (29.3) | 0.102 |
| Metastasis, n (%) | 273 (78.7) | 82 (70.7) | |
| Age at diagnosis (years), median (range) | 58 (15–90) | 60 (20–90) | 0.126 |
| Breslov thickness (mm), median (range) | 3 (0.25–15.55) | 4 (0–17.75) | 0.374 |
| Clark level, n (%) | |||
| I | 0 | 1 (0.01) | 0.229 |
| II | 15 (0.06) | 3 (0.03) | |
| III | 60 (25.9) | 17 (19.8) | |
| IV | 120 (51.7) | 47 (54.4) | |
| V | 37 (16.0) | 18 (20.9) | |
| Ulceration (present), n (%) | 118 (50.9) | 51 (60.7) | 0.182 |
| BRAF mutant, yes, n (%) | 149 (54.2) | 34 (42.0) | 0.023 |
| NRAS mutant, yes, n (%) | 75 (27.3) | 19 (23.5) | 0.422 |
| NF1 mutant, yes, n (%) | 30 (10.9) | 15 (18.5) | 0.034 |
| FGA, ave (SD) | 0.27 (0–0.77) | 0.33 (0–0.97) | 1.46 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czerwinska, P.; Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.A. Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein. Cancers 2020, 12, 2998. https://doi.org/10.3390/cancers12102998
Czerwinska P, Jaworska AM, Wlodarczyk NA, Mackiewicz AA. Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein. Cancers. 2020; 12(10):2998. https://doi.org/10.3390/cancers12102998
Chicago/Turabian StyleCzerwinska, Patrycja, Anna Maria Jaworska, Nikola Agata Wlodarczyk, and Andrzej Adam Mackiewicz. 2020. "Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein" Cancers 12, no. 10: 2998. https://doi.org/10.3390/cancers12102998
APA StyleCzerwinska, P., Jaworska, A. M., Wlodarczyk, N. A., & Mackiewicz, A. A. (2020). Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein. Cancers, 12(10), 2998. https://doi.org/10.3390/cancers12102998