The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma

 ,
, 
Simple Summary
Abstract
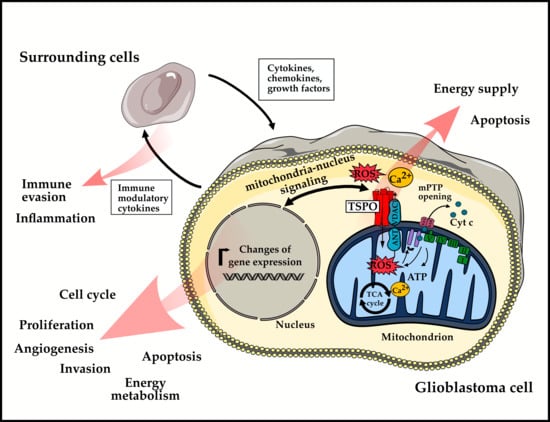
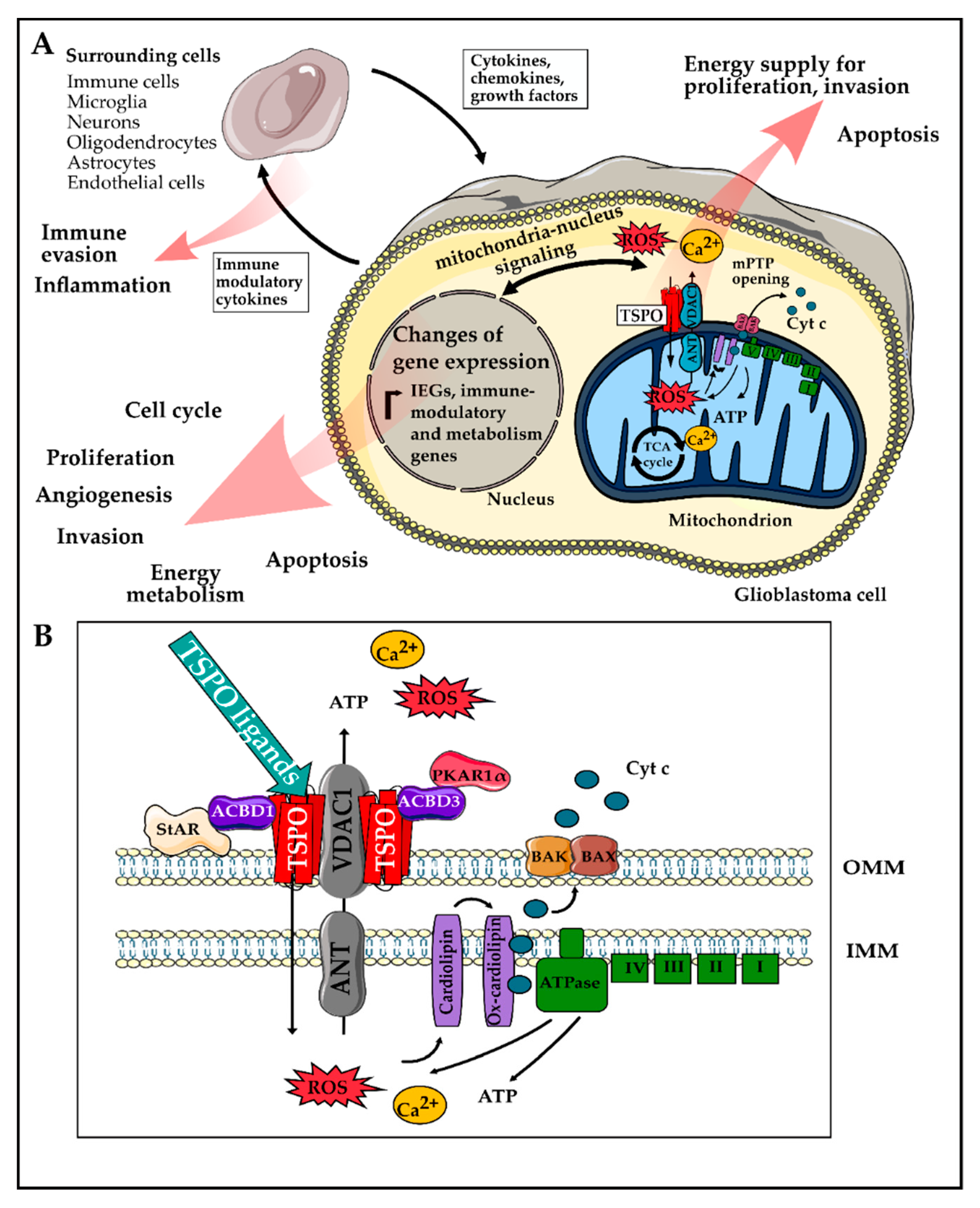
1. Introduction
1.1. Glioblastoma Pathophysiology
1.2. Translocator Protein TSPO
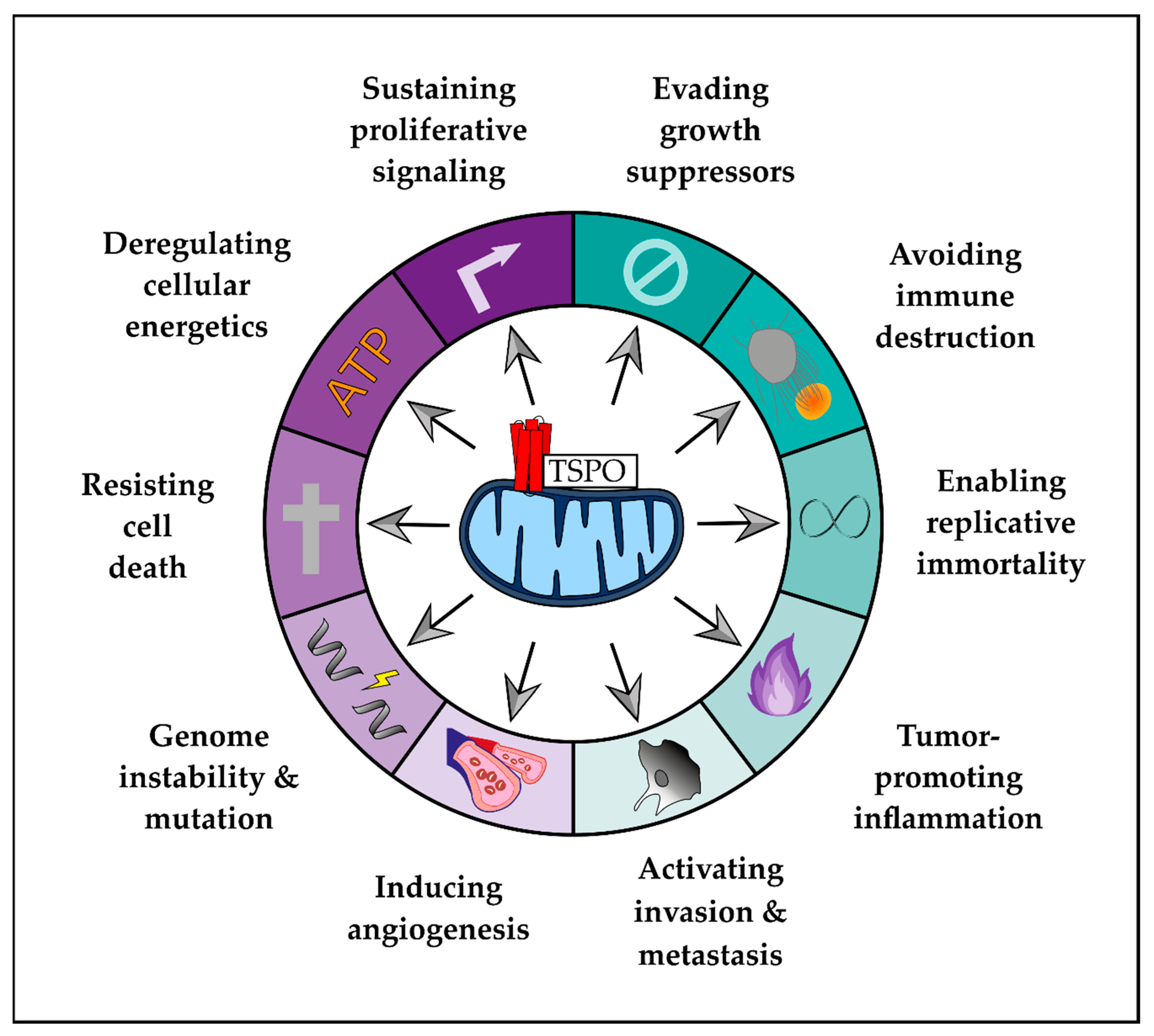
2. Role of TSPO in Hallmarks of GBM
2.1. Enabling Characteristics
2.1.1. Genome Instability and Mutation
2.1.2. Tumor Promoting Inflammation
2.2. Hallmarks of Cancer
2.2.1. Sustaining Proliferative Signaling
2.2.2. Evading Growth Suppressors
2.2.3. Resisting Cell Death
2.2.4. Enabling Replicative Immortality
2.2.5. Inducing Angiogenesis
2.2.6. Activating Invasion and Metastasis
2.3. Emerging Hallmarks
2.3.1. Evading Immune Destruction
2.3.2. Reprogramming Energy Metabolism
3. In Vivo-Monitoring of TSPO
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Tso, C.-L.; Freije, W.A.; Day, A.; Chen, Z.; Merriman, B.; Perlina, A.; Lee, Y.; Dia, E.Q.; Yoshimoto, K.; Mischel, P.S.; et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006, 66, 159–167. [Google Scholar] [CrossRef]
- Li, C.; Wang, S.; Yan, J.-L.; Piper, R.J.; Liu, H.; Torheim, T.; Kim, H.; Zou, J.; Boonzaier, N.R.; Sinha, R.; et al. Intratumoral Heterogeneity of Glioblastoma Infiltration Revealed by Joint Histogram Analysis of Diffusion Tensor Imaging. Neurosurgery 2019, 85, 524–534. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef]
- Vitorino, P.; Meyer, T. Modular control of endothelial sheet migration. Genes Dev. 2008, 22, 3268–3281. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef]
- Michaelsen, S.R.; Christensen, I.J.; Grunnet, K.; Stockhausen, M.-T.; Broholm, H.; Kosteljanetz, M.; Poulsen, H.S. Clinical variables serve as prognostic factors in a model for survival from glioblastoma multiforme: An observational study ofa cohort of consecutive non-selected patients from a single institution. BMC Cancer 2013, 13, 402. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.A.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-Oncology 2013, 15, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Bralten, L.B.C.; Kloosterhof, N.K.; Balvers, R.; Sacchetti, A.; Lapre, L.; Lamfers, M.; Leenstra, S.; de Jonge, H.; Kros, J.M.; Jansen, E.E.W.; et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann. Neurol. 2011, 69, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Yao, Y.; Xu, H.-Z.; Qin, Z.-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients with Glioblastomas. Medicine 2016, 95, e2583. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Giampazolias, E.; Tait, S.W.G. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Wu, L.-P.; Gong, Z.-F.; Wang, H.; Zhou, Z.-S.; Zhang, M.-M.; Liu, C.; Ren, H.-M.; Yang, J.; Han, Y.; Zeng, C.-Y. TSPO ligands prevent the proliferation of vascular smooth muscle cells and attenuate neointima formation through AMPK activation. Acta Pharmacol. Sin. 2020, 41, 34–46. [Google Scholar] [CrossRef]
- Veenman, L.; Levin, E.; Weisinger, G.; Leschiner, S.; Spanier, I.; Snyder, S.H.; Weizman, A.; Gavish, M. Peripheral-type benzodiazepine receptor density and in vitro tumorigenicity of glioma cell lines. Biochem. Pharmacol. 2004, 68, 689–698. [Google Scholar] [CrossRef]
- Lin, R.; Angelin, A.; Da Settimo, F.; Martini, C.; Taliani, S.; Zhu, S.; Wallace, D.C. Genetic analysis of dTSPO, an outer mitochondrial membrane protein, reveals its functions in apoptosis, longevity, and Ab42-induced neurodegeneration. Aging Cell 2014, 13, 507–518. [Google Scholar] [CrossRef]
- Zeno, S.; Zaaroor, M.; Leschiner, S.; Veenman, L.; Gavish, M. CoCl(2) induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry 2009, 48, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Kugler, W.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Lakomek, M.; Gavish, M. Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell. Oncol. 2008, 30, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gallo, K.A. The 18-kDa translocator protein (TSPO) disrupts mammary epithelial morphogenesis and promotes breast cancer cell migration. PLoS ONE 2013, 8, e71258. [Google Scholar] [CrossRef] [PubMed]
- Lejri, I.; Grimm, A.; Hallé, F.; Abarghaz, M.; Klein, C.; Maitre, M.; Schmitt, M.; Bourguignon, J.-J.; Mensah-Nyagan, A.G.; Bihel, F.; et al. TSPO Ligands Boost Mitochondrial Function and Pregnenolone Synthesis. J. Alzheimer’s Dis. 2019, 72, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Slim, D.; Bader, S.; Koch, V.; Heinl, E.-S.; Alvarez-Carbonell, D.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019, 20, 3359. [Google Scholar] [CrossRef] [PubMed]
- Zeno, S.; Veenman, L.; Katz, Y.; Bode, J.; Gavish, M.; Zaaroor, M. The 18 kDa mitochondrial translocator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygen species (ROS). Curr. Mol. Med. 2012, 12, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, H.; Kononen, J.; Haapasalo, H.; Helén, P.; Sallinen, P.; Harjuntausta, T.; Helin, H.; Alho, H. Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human astrocytomas: Relationship to cell proliferation. Cancer Res. 1995, 55, 2691–2695. [Google Scholar]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. Neurooncol. 2007, 81, 1–7. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Kanu, O.O.; Hughes, B.; Di, C.; Lin, N.; Fu, J.; Bigner, D.D.; Yan, H.; Adamson, C. Glioblastoma Multiforme Oncogenomics and Signaling Pathways. Clin. Med. Oncol. 2009, 3, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Brennan, C.; Wen, P.Y.; Durso, L.; Ligon, K.L.; Richardson, A.; Khatry, D.; Feng, B.; Sinha, R.; Louis, D.N.; et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 2006, 66, 11502–11513. [Google Scholar] [CrossRef] [PubMed]
- van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.-K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef]
- Ahmed, R.; Oborski, M.J.; Hwang, M.; Lieberman, F.S.; Mountz, J.M. Malignant gliomas: Current perspectives in diagnosis, treatment, and early response assessment using advanced quantitative imaging methods. Cancer Manag. Res. 2014, 6, 149–170. [Google Scholar] [CrossRef]
- Kleihues, P.; Ohgaki, H. Primary and secondary glioblastomas: From concept to clinical diagnosis. Neuro-Oncology 1999, 1, 44–51. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.M.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Louis, D.N.; Weller, M.; Hau, P. Refined brain tumor diagnostics and stratified therapies: The requirement for a multidisciplinary approach. Acta Neuropathol. 2013, 126, 21–37. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Stifani, S. NF-κB Signalling in Glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, N.; Nakatani, Y.; Yamashita, D.; Ohue, S.; Ohnishi, T.; Kondo, T. Eva1 Maintains the Stem-like Character of Glioblastoma-Initiating Cells by Activating the Noncanonical NF-κB Signaling Pathway. Cancer Res. 2016, 76, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Shimamura, T.; Barbera, S.; Bejcek, B.E. NF-kappaB controls growth of glioblastomas/astrocytomas. Mol. Cell. Biochem. 2008, 307, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Ezhilarasan, R.; Phillips, E.; Gallego-Perez, D.; Sparks, A.; Taylor, D.; Ladner, K.; Furuta, T.; Sabit, H.; Chhipa, R.; et al. Serine/Threonine Kinase MLK4 Determines Mesenchymal Identity in Glioma Stem Cells in an NF-κB-dependent Manner. Cancer Cell 2016, 29, 201–213. [Google Scholar] [CrossRef]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, M.; Zuo, J.; Wahafu, A.; Mao, P.; Li, R.; Wu, W.; Xie, W.; Wang, J. Nuclear factor I A promotes temozolomide resistance in glioblastoma via activation of nuclear factor κB pathway. Life Sci. 2019, 236, 116917. [Google Scholar] [CrossRef]
- Gray, G.K.; McFarland, B.C.; Nozell, S.E.; Benveniste, E.N. NF-κB and STAT3 in glioblastoma: Therapeutic targets coming of age. Expert Rev. Neurother. 2014, 14, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, Ł.; Jaipuria, G.; Becker, S.; Zweckstetter, M. Structure of the mammalian TSPO/PBR protein. Biochem. Soc. Trans. 2015, 43, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Delavoie, F.; Li, H.; Hardwick, M.; Robert, J.-C.; Giatzakis, C.; Péranzi, G.; Yao, Z.-X.; Maccario, J.; Lacapère, J.-J.; Papadopoulos, V. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: Functional significance in drug ligand and cholesterol binding. Biochemistry 2003, 42, 4506–4519. [Google Scholar] [CrossRef] [PubMed]
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar] [PubMed]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.-J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.-R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Verma, A.; Nye, J.S.; Snyder, S.H. Porphyrins are endogenous ligands for the mitochondrial (peripheral-type) benzodiazepine receptor. Proc. Natl. Acad. Sci. USA 1987, 84, 2256–2260. [Google Scholar] [CrossRef]
- Guidotti, A.; Forchetti, C.M.; Corda, M.G.; Konkel, D.; Bennett, C.D.; Costa, E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc. Natl. Acad. Sci. USA 1983, 80, 3531–3535. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Qiu, Z.-K.; Chen, X.-F.; Zhao, N.; Chen, H.-X.; Xue, R.; Zhang, Y.-Z.; Yang, R.-F.; Li, Y.-F. Involvement of allopregnanolone in the anti-PTSD-like effects of AC-5216. J. Psychopharmacol. (Oxford) 2016, 30, 474–481. [Google Scholar] [CrossRef]
- Batarseh, A.; Barlow, K.D.; Martinez-Arguelles, D.B.; Papadopoulos, V. Functional characterization of the human translocator protein (18kDa) gene promoter in human breast cancer cell lines. Biochim. Biophys. Acta 2012, 1819, 38–56. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, C.; Görg, B.; Keitel, V.; Pirev, E.; Kröncke, K.D.; Schliess, F.; Häussinger, D. Hypoosmotic swelling affects zinc homeostasis in cultured rat astrocytes. Glia 2009, 57, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, A.; Li, J.; Papadopoulos, V. Protein kinase C epsilon regulation of translocator protein (18 kDa) Tspo gene expression is mediated through a MAPK pathway targeting STAT3 and c-Jun transcription factors. Biochemistry 2010, 49, 4766–4778. [Google Scholar] [CrossRef]
- Hardwick, M.; Cavalli, L.R.; Barlow, K.D.; Haddad, B.R.; Papadopoulos, V. Peripheral-type benzodiazepine receptor (PBR) gene amplification in MDA-MB-231 aggressive breast cancer cells. Cancer Genet. Cytogenet. 2002, 139, 48–51. [Google Scholar] [CrossRef]
- Han, Z.; Slack, R.S.; Li, W.; Papadopoulos, V. Expression of peripheral benzodiazepine receptor (PBR) in human tumors: Relationship to breast, colorectal, and prostate tumor progression. J. Recept. Signal Transduct. Res. 2003, 23, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Kirchleitner, S.V.; Zhao, D.; Li, M.; Tonn, J.-C.; Glass, R.; Kälin, R.E. Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells. Int. J. Mol. Sci. 2020, 21, 612. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef]
- de Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, Y.; Soto-Reyes, E.; Garcia-Cuellar, C.M.; Cacho-Diaz, B.; Santamaria, A.; Rangel-Lopez, E. Role of Epigenetics and Oxidative Stress in Gliomagenesis. CNS Neurol. Disord. Drug Targets 2017, 16, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Panickar, K.S.; Norenberg, M.D. Effects on free radical generation by ligands of the peripheral benzodiazepine receptor in cultured neural cells. J. Neurochem. 2002, 83, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, A.; Giatzakis, C.; Papadopoulos, V. Phorbol-12-myristate 13-acetate acting through protein kinase Cepsilon induces translocator protein (18-kDa) TSPO gene expression. Biochemistry 2008, 47, 12886–12899. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; Campanella, M. TSPO is a REDOX regulator of cell mitophagy. Biochem. Soc. Trans. 2015, 43, 543–552. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Grimm, E.; Heimberger, A.B. Innate immune functions of microglia isolated from human glioma patients. J. Transl. Med. 2006, 4, 15. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Morisse, M.C.; Jouannet, S.; Dominguez-Villar, M.; Sanson, M.; Idbaih, A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: Unraveling promising targeted therapies. Expert Rev. Neurother. 2018, 18, 729–737. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. (Landmark Ed) 2017, 22, 1805–1829. [Google Scholar] [CrossRef] [PubMed]
- Vivash, L.; O’Brien, T.J. Imaging Microglial Activation with TSPO PET: Lighting Up Neurologic Diseases? J. Nucl. Med. 2016, 57, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, S.; Li, X.; Yang, B.; Wu, C. The ligands of translocator protein inhibit human Th1 responses and the rejection of murine skin allografts. Clin. Sci. 2017, 131, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-Y.; Yu, J.-Z.; Li, Q.-Y.; Ma, C.-G.; Lu, C.-Z.; Xiao, B.-G. TSPO-specific ligand vinpocetine exerts a neuroprotective effect by suppressing microglial inflammation. Neuron Glia Biol. 2011, 7, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.B.; Khoo, C.; Ryu, J.K.; van Breemen, E.; Kim, S.U.; McLarnon, J.G. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and Ca2+i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J. Neurochem. 2002, 83, 546–555. [Google Scholar] [CrossRef]
- Choi, J.; Ifuku, M.; Noda, M.; Guilarte, T.R. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 2011, 59, 219–230. [Google Scholar] [CrossRef]
- Barron, A.M.; Garcia-Segura, L.M.; Caruso, D.; Jayaraman, A.; Lee, J.-W.; Melcangi, R.C.; Pike, C.J. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 8891–8897. [Google Scholar] [CrossRef]
- Beckers, L.; Ory, D.; Geric, I.; Declercq, L.; Koole, M.; Kassiou, M.; Bormans, G.; Baes, M. Increased Expression of Translocator Protein (TSPO) Marks Pro-inflammatory Microglia but Does Not Predict Neurodegeneration. Mol. Imaging Biol. 2018, 20, 94–102. [Google Scholar] [CrossRef]
- Pannell, M.; Economopoulos, V.; Wilson, T.C.; Kersemans, V.; Isenegger, P.G.; Larkin, J.R.; Smart, S.; Gilchrist, S.; Gouverneur, V.; Sibson, N.R. Imaging of translocator protein upregulation is selective for pro-inflammatory polarized astrocytes and microglia. Glia 2020, 68, 280–297. [Google Scholar] [CrossRef]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, E.D.; Tremolanti, C.; Costa, B.; Giacomelli, C.; Milenkovic, V.M.; Bader, S.; Wetzel, C.H.; Rupprecht, R.; Taliani, S.; Settimo, F.D.; et al. Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. Int. J. Mol. Sci. 2019, 20, 4467. [Google Scholar] [CrossRef] [PubMed]
- Doucette, T.; Rao, G.; Rao, A.; Shen, L.; Aldape, K.; Wei, J.; Dziurzynski, K.; Gilbert, M.; Heimberger, A.B. Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol. Res. 2013, 1, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hon, G.C.; Villa, G.R.; Turner, K.M.; Ikegami, S.; Yang, H.; Ye, Z.; Li, B.; Kuan, S.; Lee, A.Y.; et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol. Cell 2015, 60, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Betensky, R.A.; Pasedag, S.M.; Louis, D.N. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006, 66, 5618–5623. [Google Scholar] [CrossRef]
- Brown, R. Location-dependent role of the human glioma cell peripheral-type benzodiazepine receptor in proliferation and steroid biosynthesis. Cancer Lett. 2000, 156, 125–132. [Google Scholar] [CrossRef]
- Rechichi, M.; Salvetti, A.; Chelli, B.; Costa, B.; Da Pozzo, E.; Spinetti, F.; Lena, A.; Evangelista, M.; Rainaldi, G.; Martini, C.; et al. TSPO over-expression increases motility, transmigration and proliferation properties of C6 rat glioma cells. Biochim. Biophys. Acta 2008, 1782, 118–125. [Google Scholar] [CrossRef]
- Liu, G.-J.; Middleton, R.J.; Kam, W.W.-Y.; Chin, D.Y.; Hatty, C.R.; Chan, R.H.Y.; Banati, R.B. Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 2017, 16, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.; Veenman, L.; Caballero, B.; Lakomek, M.; Kugler, W.; Gavish, M. The 18 kDa translocator protein influences angiogenesis, as well as aggressiveness, adhesion, migration, and proliferation of glioblastoma cells. Pharmacogenet. Genom. 2012, 22, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, D.; Wang, H.; Cai, M.; Li, C.; Zhang, X.; Chen, H.; Hu, Y.; Zhang, X.; Ying, M.; et al. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis, and a growth-promoting metabolic shift toward glycolysis in glioblastoma. Neuro-Oncology 2020, 22, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Cosimelli, B.; Simorini, F.; Taliani, S.; La Motta, C.; Da Settimo, F.; Severi, E.; Greco, G.; Novellino, E.; Costa, B.; Da Pozzo, E.; et al. Tertiary amides with a five-membered heteroaromatic ring as new probes for the translocator protein. Eur. J. Med. Chem. 2011, 46, 4506–4520. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Ferraz-de-Paula, V.; Pinheiro, M.L.; Ribeiro, A.; Quinteiro-Filho, W.M.; Rone, M.B.; Martinez-Arguelles, D.B.; Dagli, M.L.Z.; Papadopoulos, V.; Palermo-Neto, J. Translocator protein (18 kDa) mediates the pro-growth effects of diazepam on Ehrlich tumor cells in vivo. Eur. J. Pharmacol. 2010, 626, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef]
- Gavish, M.; Veenman, L. Regulation of Mitochondrial, Cellular, and Organismal Functions by TSPO. Adv. Pharmacol. 2018, 82, 103–136. [Google Scholar] [CrossRef]
- Kabat, G.C.; Etgen, A.M.; Rohan, T.E. Do steroid hormones play a role in the etiology of glioma? Cancer Epidemiol. Biomark. Prev. 2010, 19, 2421–2427. [Google Scholar] [CrossRef]
- Batistatou, A.; Kyzas, P.A.; Goussia, A.; Arkoumani, E.; Voulgaris, S.; Polyzoidis, K.; Agnantis, N.J.; Stefanou, D. Estrogen receptor beta (ERbeta) protein expression correlates with BAG-1 and prognosis in brain glial tumours. J. Neurooncol. 2006, 77, 17–23. [Google Scholar] [CrossRef]
- Yague, J.G.; Lavaque, E.; Carretero, J.; Azcoitia, I.; Garcia-Segura, L.M. Aromatase, the enzyme responsible for estrogen biosynthesis, is expressed by human and rat glioblastomas. Neurosci. Lett. 2004, 368, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sareddy, G.R.; Zhou, M.; Viswanadhapalli, S.; Li, X.; Lai, Z.; Tekmal, R.R.; Brenner, A.; Vadlamudi, R.K. Differential Effects of Estrogen Receptor β Isoforms on Glioblastoma Progression. Cancer Res. 2018, 78, 3176–3189. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lozano, D.C.; Piña-Medina, A.G.; Hansberg-Pastor, V.; Bello-Alvarez, C.; Camacho-Arroyo, I. Testosterone Promotes Glioblastoma Cell Proliferation, Migration, and Invasion Through Androgen Receptor Activation. Front. Endocrinol. (Lausanne) 2019, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jiang, Y.; Wei, W.; Cong, P.; Ding, Y.; Xiang, L.; Wu, K. Androgen receptor signaling regulates growth of glioblastoma multiforme in men. Tumour Biol. 2015, 36, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Cheng, C.; Lan, X.; Xing, R.; Chen, Z.; Zhao, H.; Sun, J.; Wang, Y.; Niu, C.; Zhang, B.; et al. Regulation of p53wt glioma cell proliferation by androgen receptor-mediated inhibition of small VCP/p97-interacting protein expression. Oncotarget 2017, 8, 23142–23154. [Google Scholar] [CrossRef]
- Zalcman, N.; Canello, T.; Ovadia, H.; Charbit, H.; Zelikovitch, B.; Mordechai, A.; Fellig, Y.; Rabani, S.; Shahar, T.; Lossos, A.; et al. Androgen receptor: A potential therapeutic target for glioblastoma. Oncotarget 2018, 9, 19980–19993. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Büschges, R.; Wolter, M.; Reifenberger, J.; Boström, J.; Kraus, J.A.; Schlegel, U.; Reifenberger, G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999, 59, 6091–6096. [Google Scholar]
- Riemenschneider, M.J.; Knobbe, C.B.; Reifenberger, G. Refined mapping of 1q32 amplicons in malignant gliomas confirms MDM4 as the main amplification target. Int. J. Cancer 2003, 104, 752–757. [Google Scholar] [CrossRef]
- Maaser, K.; Sutter, A.P.; Krahn, A.; Höpfner, M.; Grabowski, P.; Scherübl, H. Cell cycle-related signaling pathways modulated by peripheral benzodiazepine receptor ligands in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2004, 324, 878–886. [Google Scholar] [CrossRef]
- Maaser, K.; Höpfner, M.; Jansen, A.; Weisinger, G.; Gavish, M.; Kozikowski, A.P.; Weizman, A.; Carayon, P.; Riecken, E.O.; Zeitz, M.; et al. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human colorectal cancer cells. Br. J. Cancer 2001, 85, 1771–1780. [Google Scholar] [CrossRef]
- Carmel, I.; Fares, F.A.; Leschiner, S.; Scherübl, H.; Weisinger, G.; Gavish, M. Peripheral-type benzodiazepine receptors in the regulation of proliferation of MCF-7 human breast carcinoma cell line. Biochem. Pharmacol. 1999, 58, 273–278. [Google Scholar] [CrossRef]
- Liu, X.; Yang, J.; Zhang, Y.; Fang, Y.; Wang, F.; Wang, J.; Zheng, X.; Yang, J. A systematic study on drug-response associated genes using baseline gene expressions of the Cancer Cell Line Encyclopedia. Sci. Rep. 2016, 6, 22811. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Wei, B.; Wang, Z.; Kong, D.; Gao, Z.; Miao, Z. Bioinformatics analysis of the CDK2 functions in neuroblastoma. Mol. Med. Rep. 2018, 17, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.; Veenman, L.; Vainshtein, A.; Kugler, W.; Rosenberg, N.; Gavish, M. Modulation of Gene Expression Associated with the Cell Cycle and Tumorigenicity of Glioblastoma Cells by the 18 kDa Translocator Protein (TSPO). Austin J. Pharmacol. Ther. 2014, 14, 1053. [Google Scholar]
- Rosenberg, N.; Rosenberg, O.; Leschiner, S.; Soudry, M.; Weizman, A.; Veenman, L.; Gavish, M. 7 Translocator protein 18kDa (TSPO) endogenous ligand affect metabolic activity and cell cycle of human osteoblast-like cell. Mitochondrion 2007, 7, 406. [Google Scholar] [CrossRef]
- Mendonça-Torres, M.C.; Roberts, S.S. The translocator protein (TSPO) ligand PK11195 induces apoptosis and cell cycle arrest and sensitizes to chemotherapy treatment in pre- and post-relapse neuroblastoma cell lines. Cancer Biol. Ther. 2013, 14, 319–326. [Google Scholar] [CrossRef]
- Yasin, N.; Veenman, L.; Singh, S.; Azrad, M.; Bode, J.; Vainshtein, A.; Caballero, B.; Marek, I.; Gavish, M. Classical and Novel TSPO Ligands for the Mitochondrial TSPO Can Modulate Nuclear Gene Expression: Implications for Mitochondrial Retrograde Signaling. Int. J. Mol. Sci. 2017, 18, 786. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Yan, S.-F.; Xue, H.; Zhang, P.; Sun, J.-T.; Li, G. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 10607–10619. [Google Scholar] [CrossRef]
- Raza, S.M.; Lang, F.F.; Aggarwal, B.B.; Fuller, G.N.; Wildrick, D.M.; Sawaya, R. Necrosis and glioblastoma: A friend or a foe? A review and a hypothesis. Neurosurgery 2002, 51, 2–12; discussion 12–13. [Google Scholar] [CrossRef]
- Savitskaya, M.A.; Onishchenko, G.E. Mechanisms of Apoptosis. Biochem. Mosc. 2015, 80, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Levin, E.; Premkumar, A.; Veenman, L.; Kugler, W.; Leschiner, S.; Spanier, I.; Weisinger, G.; Lakomek, M.; Weizman, A.; Snyder, S.H.; et al. The peripheral-type benzodiazepine receptor and tumorigenicity: Isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry 2005, 44, 9924–9935. [Google Scholar] [CrossRef] [PubMed]
- Shoukrun, R.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Karry, R.; Katz, Y.; Weisinger, G.; Weizman, A.; Gavish, M. The 18-kDa translocator protein, formerly known as the peripheral-type benzodiazepine receptor, confers proapoptotic and antineoplastic effects in a human colorectal cancer cell line. Pharmacogenet. Genom. 2008, 18, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Bode, J.; Gaitner, M.; Caballero, B.; Pe’er, Y.; Zeno, S.; Kietz, S.; Kugler, W.; Lakomek, M.; Gavish, M. Effects of 18-kDa translocator protein knockdown on gene expression of glutamate receptors, transporters, and metabolism, and on cell viability affected by glutamate. Pharmacogenet. Genom. 2012, 22, 606–619. [Google Scholar] [CrossRef]
- Santidrián, A.F.; Cosialls, A.M.; Coll-Mulet, L.; Iglesias-Serret, D.; de Frias, M.; González-Gironès, D.M.; Campàs, C.; Domingo, A.; Pons, G.; Gil, J. The potential anticancer agent PK11195 induces apoptosis irrespective of p53 and ATM status in chronic lymphocytic leukemia cells. Haematologica 2007, 92, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Taliani, S.; Da Pozzo, E.; Giacomelli, C.; Costa, B.; Trincavelli, M.L.; Rossi, L.; La Pietra, V.; Barresi, E.; Carotenuto, A.; et al. Apoptosis therapy in cancer: The first single-molecule co-activating p53 and the translocator protein in glioblastoma. Sci. Rep. 2014, 4, 4749. [Google Scholar] [CrossRef] [PubMed]
- Castellano, S.; Taliani, S.; Viviano, M.; Milite, C.; Da Pozzo, E.; Costa, B.; Barresi, E.; Bruno, A.; Cosconati, S.; Marinelli, L.; et al. Structure-activity relationship refinement and further assessment of 4-phenylquinazoline-2-carboxamide translocator protein ligands as antiproliferative agents in human glioblastoma tumors. J. Med. Chem. 2014, 57, 2413–2428. [Google Scholar] [CrossRef]
- Chelli, B.; Salvetti, A.; Da Pozzo, E.; Rechichi, M.; Spinetti, F.; Rossi, L.; Costa, B.; Lena, A.; Rainaldi, G.; Scatena, F.; et al. PK 11195 differentially affects cell survival in human wild-type and 18 kDa translocator protein-silenced ADF astrocytoma cells. J. Cell. Biochem. 2008, 105, 712–723. [Google Scholar] [CrossRef]
- Bono, F.; Lamarche, I.; Prabonnaud, V.; Le Fur, G.; Herbert, J.M. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem. Biophys. Res. Commun. 1999, 265, 457–461. [Google Scholar] [CrossRef]
- Obame, F.N.; Zini, R.; Souktani, R.; Berdeaux, A.; Morin, D. Peripheral benzodiazepine receptor-induced myocardial protection is mediated by inhibition of mitochondrial membrane permeabilization. J. Pharmacol. Exp. Ther. 2007, 323, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Veenman, L.; Bode, J.; Leschiner, S.; Gavish, M. Concentration-dependent bimodal effect of specific 18 kDa translocator protein (TSPO) ligands on cell death processes induced by ammonium chloride: Potential implications for neuropathological effects due to hyperammonemia. CNS Neurol. Disord. Drug Targets 2014, 13, 574–592. [Google Scholar] [CrossRef] [PubMed]
- Šileikytė, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabò, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [PubMed]
- Hans, G.; Wislet-Gendebien, S.; Lallemend, F.; Robe, P.; Rogister, B.; Belachew, S.; Nguyen, L.; Malgrange, B.; Moonen, G.; Rigo, J.-M. Peripheral benzodiazepine receptor (PBR) ligand cytotoxicity unrelated to PBR expression. Biochem. Pharmacol. 2005, 69, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Polo, R.-A.; Carvalho, G.; Braun, T.; Decaudin, D.; Fabre, C.; Larochette, N.; Perfettini, J.-L.; Djavaheri-Mergny, M.; Youlyouz-Marfak, I.; Codogno, P.; et al. PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor. Oncogene 2005, 24, 7503–7513. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabò, I. The mitochondrial permeability transition. Biochim. Biophys. Acta (BBA)—Rev. Biomembr. 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Zeng, Y. Peripheral benzodiazepine receptor ligand, PK11195 induces mitochondria cytochrome c release and dissipation of mitochondria potential via induction of mitochondria permeability transition. Eur. J. Pharmacol. 2007, 560, 117–122. [Google Scholar] [CrossRef]
- Chelli, B.; Falleni, A.; Salvetti, F.; Gremigni, V.; Lucacchini, A.; Martini, C. Peripheral-type benzodiazepine receptor ligands. Biochem. Pharmacol. 2001, 61, 695–705. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Simbula, G.; Gilfor, E.; Hoek, J.B.; Farber, J.L. Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem. 1994, 269, 31041–31046. [Google Scholar]
- Azarashvili, T.; Krestinina, O.; Baburina, Y.; Odinokova, I.; Grachev, D.; Papadopoulos, V.; Akatov, V.; Lemasters, J.J.; Reiser, G. Combined effect of G3139 and TSPO ligands on Ca(2+)-induced permeability transition in rat brain mitochondria. Arch. Biochem. Biophys. 2015, 587, 70–77. [Google Scholar] [CrossRef]
- Shargorodsky, L.; Veenman, L.; Caballero, B.; Pe’er, Y.; Leschiner, S.; Bode, J.; Gavish, M. The nitric oxide donor sodium nitroprusside requires the 18 kDa Translocator Protein to induce cell death. Apoptosis 2012, 17, 647–665. [Google Scholar] [CrossRef]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wang, K.; Zirkin, B.; Papadopoulos, V. CRISPR/Cas9–Mediated Tspo Gene Mutations Lead to Reduced Mitochondrial Membrane Potential and Steroid Formation in MA-10 Mouse Tumor Leydig Cells. Endocrinology 2018, 159, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Tian, M.; Yin, S.; Lai, S.; Zhou, Y.; Chen, J.; He, M.; Liao, Z. Downregulation of TSPO expression inhibits oxidative stress and maintains mitochondrial homeostasis in cardiomyocytes subjected to anoxia/reoxygenation injury. Biomed. Pharmacother. 2020, 121, 109588. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef]
- Takano, S.; Yamashita, T.; Ohneda, O. Molecular therapeutic targets for glioma angiogenesis. J. Oncol. 2010, 2010, 351908. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef]
- Veenman, L.; Gavish, M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther. 2006, 110, 503–524. [Google Scholar] [CrossRef]
- Son, H.; Moon, A. Epithelial-mesenchymal Transition and Cell Invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Onken, J.; Moeckel, S.; Leukel, P.; Leidgens, V.; Baumann, F.; Bogdahn, U.; Vollmann-Zwerenz, A.; Hau, P. Versican isoform V1 regulates proliferation and migration in high-grade gliomas. J. Neurooncol. 2014, 120, 73–83. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Batarseh, A.; Papadopoulos, V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 2010, 327, 1–12. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.; Ryu, B.; Chi, S.-G.; Park, H. Caveolin-1 is associated with VCAM-1 dependent adhesion of gastric cancer cells to endothelial cells. Cell. Physiol. Biochem. 2006, 17, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Mäenpää, A.; Kovanen, P.E.; Paetau, A.; Jäáskeläinen, J.; Timonen, T. Lymphocyte adhesion molecule ligands and extracellular matrix proteins in gliomas and normal brain: Expression of VCAM-1 in gliomas. Acta Neuropathol. 1997, 94, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.K.; Lee, Y.R.; Lim, S.Y.; Lee, E.J.; Choi, S.; Cho, E.J.; Park, M.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012, 586, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, Y.; Hao, Y.; Ma, Q.; Dai, J.; Liang, Z.; Liu, Y.; Li, X.; Song, Y.; Si, C. Vinpocetine Inhibited the CpG Oligodeoxynucleotide-induced Immune Response in Plasmacytoid Dendritic Cells. Immunol. Investig. 2017, 46, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef]
- Dey, M.; Chang, A.L.; Miska, J.; Wainwright, D.A.; Ahmed, A.U.; Balyasnikova, I.V.; Pytel, P.; Han, Y.; Tobias, A.; Zhang, L.; et al. Dendritic Cell-Based Vaccines that Utilize Myeloid Rather than Plasmacytoid Cells Offer a Superior Survival Advantage in Malignant Glioma. J. Immunol. 2015, 195, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Wang, Y.; Wang, Y.; Zhang, H.; Shang, D.; Tan, F.; Li, Y.; Chen, X. Tumor Location and Survival Outcomes in Adult Patients with Supratentorial Glioblastoma by Levels of Toll-Like Receptor 9 Expression. World Neurosurg. 2017, 97, 279–283. [Google Scholar] [CrossRef]
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid dendritic cell in immunity and cancer. J. Neuroimmunol. 2018, 322, 63–73. [Google Scholar] [CrossRef]
- Leva, G.; Klein, C.; Benyounes, J.; Hallé, F.; Bihel, F.; Collongues, N.; de Seze, J.; Mensah-Nyagan, A.-G.; Patte-Mensah, C. The translocator protein ligand XBD173 improves clinical symptoms and neuropathological markers in the SJL/J mouse model of multiple sclerosis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 3016–3027. [Google Scholar] [CrossRef]
- Karlstetter, M.; Nothdurfter, C.; Aslanidis, A.; Moeller, K.; Horn, F.; Scholz, R.; Neumann, H.; Weber, B.H.F.; Rupprecht, R.; Langmann, T. Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J. Neuroinflamm. 2014, 11, 3. [Google Scholar] [CrossRef]
- Zavala, F.; Taupin, V.; Descamps-Latscha, B. In vivo treatment with benzodiazepines inhibits murine phagocyte oxidative metabolism and production of interleukin 1, tumor necrosis factor and interleukin-6. J. Pharmacol. Exp. Ther. 1990, 255, 442–450. [Google Scholar]
- Issop, L.; Ostuni, M.A.; Lee, S.; Laforge, M.; Péranzi, G.; Rustin, P.; Benoist, J.-F.; Estaquier, J.; Papadopoulos, V.; Lacapère, J.-J. Translocator Protein-Mediated Stabilization of Mitochondrial Architecture during Inflammation Stress in Colonic Cells. PLoS ONE 2016, 11, e0152919. [Google Scholar] [CrossRef] [PubMed]
- Dienz, O.; Rincon, M. The effects of IL-6 on CD4 T cell responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; Campanella, M. TSPO: Kaleidoscopic 18-kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem. J. 2016, 473, 107–121. [Google Scholar] [CrossRef]
- Betlazar, C.; Middleton, R.J.; Banati, R.; Liu, G.-J. The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes. Cells 2020, 9, 512. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the immune system: Signal integration, propagation and termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Seliger, C.; Hau, P. Drug Repurposing of Metabolic Agents in Malignant Glioma. Int. J. Mol. Sci. 2018, 19, 2768. [Google Scholar] [CrossRef]
- Seliger, C.; Meyer, A.-L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-β2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: A new target gleaned from an old concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology 2016, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.; Rosenberg, O.; Weizman, A.; Veenman, L.; Gavish, M. In vitro catabolic effect of protoporphyrin IX in human osteoblast-like cells: Possible role of the 18 kDa mitochondrial translocator protein. J. Bioenerg. Biomembr. 2013, 45, 333–341. [Google Scholar] [CrossRef]
- Zhao, A.H.; Tu, L.N.; Mukai, C.; Sirivelu, M.P.; Pillai, V.V.; Morohaku, K.; Cohen, R.; Selvaraj, V. Mitochondrial Translocator Protein (TSPO) Function Is Not Essential for Heme Biosynthesis. J. Biol. Chem. 2016, 291, 1591–1603. [Google Scholar] [CrossRef]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.-Y.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef]
- Grimm, A.; Lejri, I.; Hallé, F.; Schmitt, M.; Götz, J.; Bihel, F.; Eckert, A. Mitochondria modulatory effects of new TSPO ligands in a cellular model of tauopathies. J. Neuroendocrinol. 2020, 32, e12796. [Google Scholar] [CrossRef]
- Xiao, J.; Liang, D.; Zhang, H.; Liu, Y.; Li, F.; Chen, Y.-H. 4′-Chlorodiazepam, a translocator protein (18 kDa) antagonist, improves cardiac functional recovery during postischemia reperfusion in rats. Exp. Biol. Med. (Maywood) 2010, 235, 478–486. [Google Scholar] [CrossRef]
- Krestinina, O.V.; Grachev, D.E.; Odinokova, I.V.; Reiser, G.; Evtodienko, Y.V.; Azarashvili, T.S. Effect of peripheral benzodiazepine receptor (PBR/TSPO) ligands on opening of Ca2+-induced pore and phosphorylation of 3.5-kDa polypeptide in rat brain mitochondria. Biochem. Mosc. 2009, 74, 421–429. [Google Scholar] [CrossRef]
- Veenman, L.; Alten, J.; Linnemannstöns, K.; Shandalov, Y.; Zeno, S.; Lakomek, M.; Gavish, M.; Kugler, W. Potential involvement of F0F1-ATP(synth)ase and reactive oxygen species in apoptosis induction by the antineoplastic agent erucylphosphohomocholine in glioblastoma cell lines: A mechanism for induction of apoptosis via the 18 kDa mitochondrial translocator protein. Apoptosis 2010, 15, 753–768. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; de Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Giacomelli, C.; Taliani, S.; Bendinelli, S.; Barresi, E.; Da Settimo, F.; Martini, C. TSPO ligand residence time influences human glioblastoma multiforme cell death/life balance. Apoptosis 2015, 20, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Black, K.L.; Ikezaki, K.; Becker, D.P. Peripheral benzodiazepine induces morphological changes and proliferation of mitochondria in glioma cells. J. Neurosci. Res. 1991, 30, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Nickels, M.L.; Tantawy, M.N.; Buck, J.R.; Manning, H.C. Preclinical imaging evaluation of novel TSPO-PET ligand 2-(5,7-Diethyl-2-(4-(2-(18)Ffluoroethoxy)phenyl)pyrazolo1,5-apyrimidin-3-yl)-N,N-diethylacetamide ((18)FVUIIS1008) in glioma. Mol. Imaging Biol. 2014, 16, 813–820. [Google Scholar] [CrossRef]
- Awde, A.R.; Boisgard, R.; Thézé, B.; Dubois, A.; Zheng, J.; Dollé, F.; Jacobs, A.H.; Tavitian, B.; Winkeler, A. The translocator protein radioligand 18F-DPA-714 monitors antitumor effect of erufosine in a rat 9L intracranial glioma model. J. Nucl. Med. 2013, 54, 2125–2131. [Google Scholar] [CrossRef]
- Su, Z.; Herholz, K.; Gerhard, A.; Roncaroli, F.; Du Plessis, D.; Jackson, A.; Turkheimer, F.; Hinz, R. 11C-(R)PK11195 tracer kinetics in the brain of glioma patients and a comparison of two referencing approaches. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1406–1419. [Google Scholar] [CrossRef]
- Su, Z.; Roncaroli, F.; Durrenberger, P.F.; Coope, D.J.; Karabatsou, K.; Hinz, R.; Thompson, G.; Turkheimer, F.E.; Janczar, K.; Du Plessis, D.; et al. The 18-kDa mitochondrial translocator protein in human gliomas: An 11C-(R)PK11195 PET imaging and neuropathology study. J. Nucl. Med. 2015, 56, 512–517. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Fleischmann, D.F.; Lindner, S.; Vettermann, F.; Brunegraf, A.; Vomacka, L.; Brendel, M.; Wenter, V.; Wetzel, C.; et al. TSPO PET for glioma imaging using the novel ligand 18F-GE-180: First results in patients with glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-K.; Guilarte, T.R. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol. Ther. 2008, 118, 1–17. [Google Scholar] [CrossRef]
- Unterrainer, M.; Fleischmann, D.F.; Vettermann, F.; Ruf, V.; Kaiser, L.; Nelwan, D.; Lindner, S.; Brendel, M.; Wenter, V.; Stöcklein, S.; et al. TSPO PET, tumour grading and molecular genetics in histologically verified glioma: A correlative 18F-GE-180 PET study. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Zinnhardt, B.; Pigeon, H.; Thézé, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Liu, J.; Culty, M.; Papadopoulos, V. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): An emerging signaling molecule. Prog. Lipid Res. 2010, 49, 218–234. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |



{kind=link}
{kind=link}
{kind=link}
| Treatment, Ligand | Cell Line | Effect on GBM | Year | Reference |
|---|---|---|---|---|
| TSPO KO | Mouse GL261 cells Human GBM1B cells | ↑ glioma growth and angiogenesis in vivo, ↑ fragmented mitochondria, glucose uptake, lactic acid conversion, ↑ ROS, ↑ glycolysis, ↓ oxidative phosphorylation and ATP production | 2020 | [104] |
| PK11195 (25 µM) | Human U118MG cells | Changes in expression of immediate early genes and transcription factors, functional changes related to cell cycle, cell death, proliferation, migration, cell viability, inflammatory, immune response, and tumorigenesis | 2017 | [127] |
| PK11195 (25 µM), TSPO KD | Human U118MG cells | Changes in gene expression related to cell cycle, apoptosis, oxidative stress, immune response, DNA repair, adhesion | 2014 | [124] |
| Ammonium chloride + PK11196, Ro5 4864, FGN-1-27 (1 nM–100 µM) | Human U118MG cells | ↓ cell death at nanomolar concentration, ↑ mitochondrial dysfunction and cell death at micromolar concentration | 2014 | [142] |
| Quinazoline derivate compound 19 (10 nM–100 µM) | Human U343 cells | ↓ proliferation, dose-dependent, ↑ dissipation of ΔΨm | 2014 | [138] |
| irDE-MPIGA (1.25 × 10−3 nmol/L + 106 cells), PIGA (2.5 µM) irDE-MPIGA + TSPO KD | Human U87MG cells | ↓ cell viability without cell cycle arrest, ↑ ΔΨm dissipation, No effect on ATPase activity No reduction of cell viability | 2014 | [205] |
| PK11195 (25 µM), TSPO KD | Human U118MG cells | ↑ tumor growth, ↑ angiogenesis, ↑ migration, ↓ adhesion of ECM | 2012 | [103] |
| Sodium nitroprusside + PK11195 (25 µM), TSPO KD | Human U118MG cells | ↓ cell death and collapse of ΔΨm, restoration of metabolic activity | 2012 | [151] |
| TSPO KD Glutamate + TSPO KD | Human U118MG cells, Rat C6 cells | Changes in glutamate metabolism ↓ DNA fragmentation | 2012 | [135] |
| PPIX (1–30 µM) + light + TSPO KD GSH + PPIX | Human U118MG cells | ↑ cell death, ↑ PPIX accumulation in cell and mitochondria ↑ PPIX accumulation in mitochondria | 2012 | [26] |
| Oxazolacetamide compound 6d (30 times of Ki value) | Human U87MG cells | ↑ cell proliferation/viability, ↑ dissipation of ΔΨm, | 2011 | [105] |
| CoCl2 + PK11195 (25 µM), TSPO KD | Human U118MG cells | ↓ cell death, ↓ ΔΨm collapse, ↓ ROS generation, ↓ cardiolipin oxidation | 2009 | [21] |
| ErPC3 + PK 11195, Ro5 4864 (25 µM–100 µM) | Human U87MG cells Human A172 cells Human U118MG cells | ↓ ErPC3 induced apoptosis, ↓ cytochrome c release, processing of caspase 9 and 3 | 2008 | [22] |
| TSPO overexpression PK11195 (1–100 µM) + TSPO overexpression | Rat C6 cells | ↑ proliferation, ↑ migration and transmigrative capabilities, ↑ anti-proliferative effect | 2007 | [100] |
| Ro5 4864 (10 nM) | Rat C6 cells, human T89G cells | ↓ cell death | 2004 | [19] |
| PK11195, Ro5 4864 (10 nM) | Rat C6 cells, human T98G cells | ↑ mitochondrial replication, shift of mitochondria from peripheral cytoplasm to the perinuclear region | 1991 | [206] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammer, L.-M.; Vollmann-Zwerenz, A.; Ruf, V.; Wetzel, C.H.; Riemenschneider, M.J.; Albert, N.L.; Beckhove, P.; Hau, P. The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma. Cancers 2020, 12, 2973. https://doi.org/10.3390/cancers12102973
Ammer L-M, Vollmann-Zwerenz A, Ruf V, Wetzel CH, Riemenschneider MJ, Albert NL, Beckhove P, Hau P. The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma. Cancers. 2020; 12(10):2973. https://doi.org/10.3390/cancers12102973
Chicago/Turabian StyleAmmer, Laura-Marie, Arabel Vollmann-Zwerenz, Viktoria Ruf, Christian H. Wetzel, Markus J. Riemenschneider, Nathalie L. Albert, Philipp Beckhove, and Peter Hau. 2020. "The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma" Cancers 12, no. 10: 2973. https://doi.org/10.3390/cancers12102973
APA StyleAmmer, L.-M., Vollmann-Zwerenz, A., Ruf, V., Wetzel, C. H., Riemenschneider, M. J., Albert, N. L., Beckhove, P., & Hau, P. (2020). The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma. Cancers, 12(10), 2973. https://doi.org/10.3390/cancers12102973