CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor




 , ,
, ,  and
and 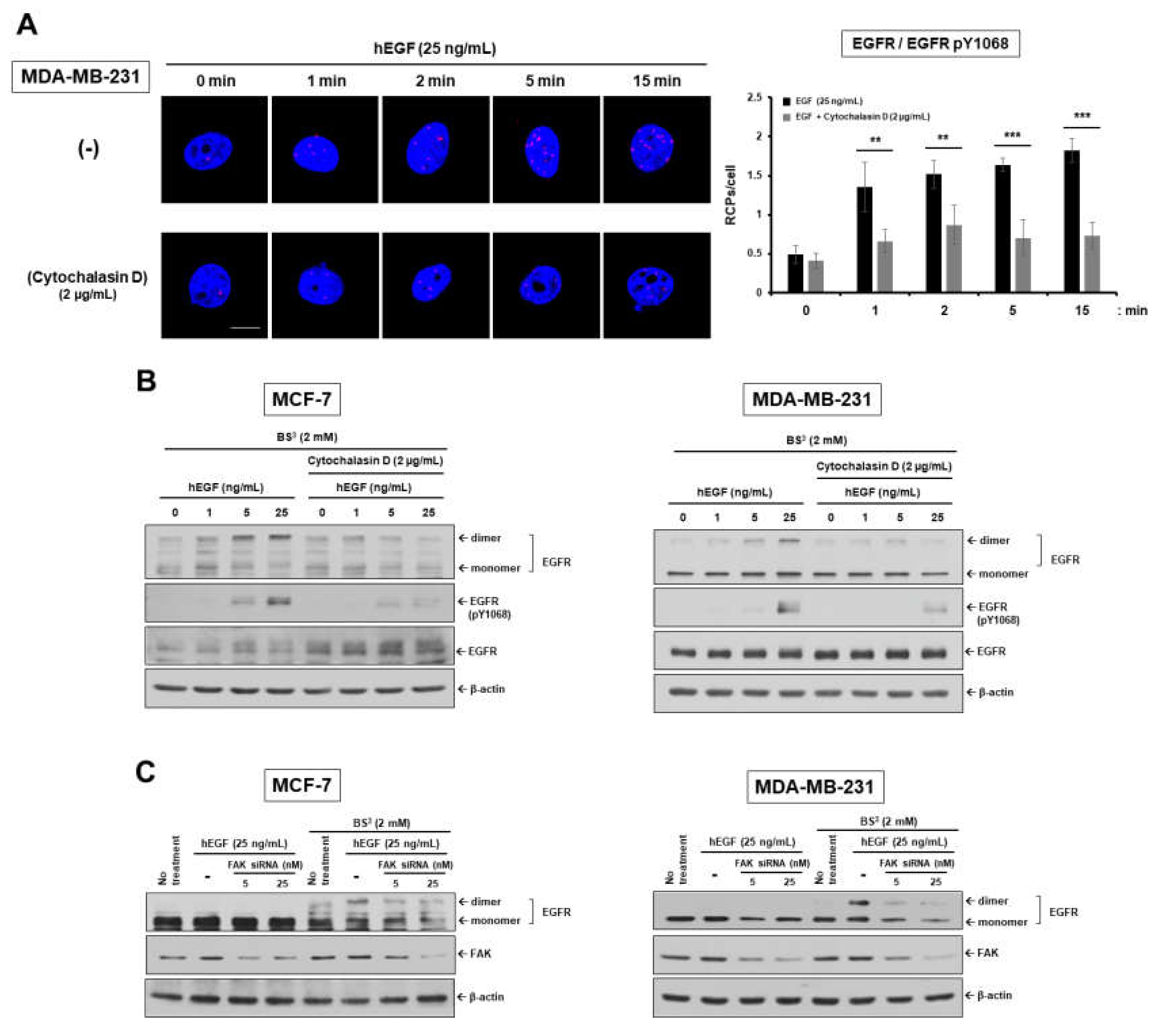{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Epidermal Growth Factor Stimulates Dimerization and Activation of the Epidermal Growth Factor Receptor through c-Src/FAK-Mediated Actin Cytoskeleton Remodeling
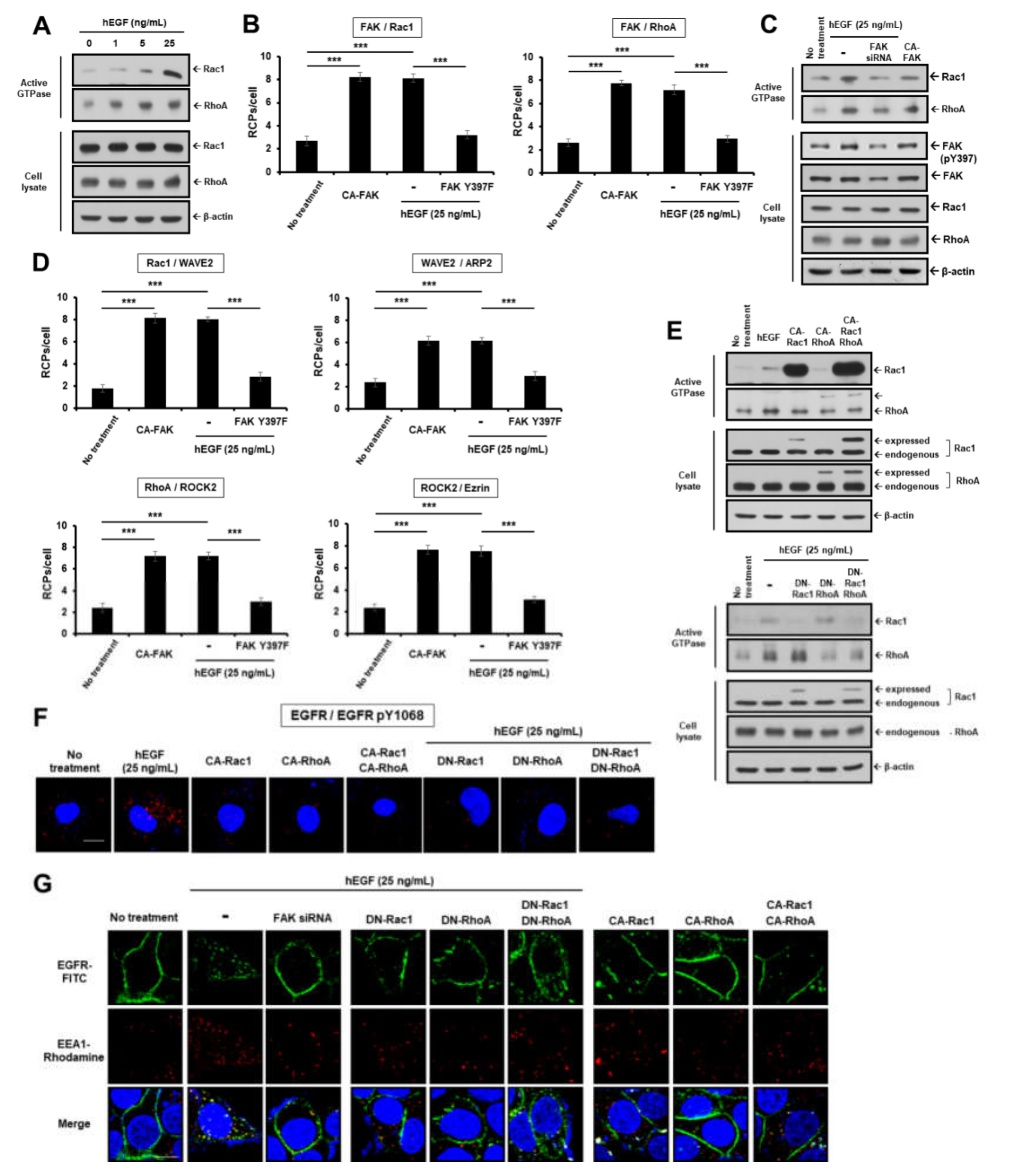
2.2. EGF Induces EGFR Dimerization and Endocytosis through FAK-Mediated RhoA and Rac1 Signaling
2.3. CD99 Activation Attenuates EGF-Induced Dimerization and Activation of EGFR via the PKA/SHP2/HRAS/PTPN12/FAK Signaling Pathway
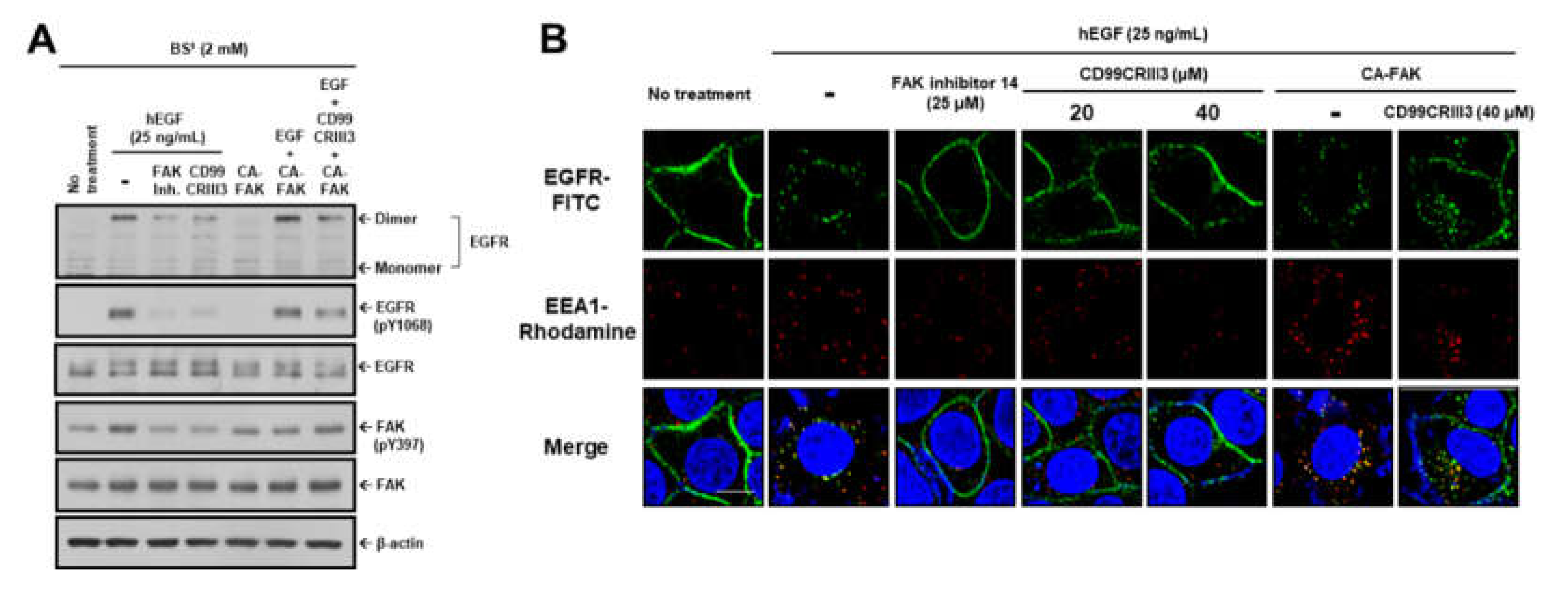
2.4. CD99CRIII3-Activated PTPN12 Regulates the Activation of EGFR Signaling through the PTPN12/FAK/ Rho/Rac Axis
2.5. CD99CRIII3 Dose-Dependently Inhibited TNBC Progression In Vivo through PTPN12-Mediated Suppression of Breast Cancer Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Synthesis of Polypeptides
4.4. Plasmids and RNA Interference
4.5. In Situ Proximity Ligation Assay (PLA)
4.6. Dimerization Assay
4.7. Active GTPase Detection
4.8. Western Blot Analysis and Immunoprecipitation
4.9. Immunofluorescence Assay (IFA)
4.10. Proliferation Assay
4.11. Tumor Xenograft and Immunohistochemistry (IHC)
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freed, D.M.; Alvarado, D.; Lemmon, M.A. Ligand regulation of a constitutively dimeric EGF receptor. Nat. Commun 2015, 6, 7380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Villeneuve, G.; Wang, Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005, 6, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 2016, 5. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef]
- Maruyama, I.N. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model”. Bioessays 2015, 37, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.; Nievergall, E.; Gegenbauer, K.; Llerena, C.; Atapattu, L.; Halle, M.; Tremblay, M.L.; Janes, P.W.; Lackmann, M. PTP-PEST controls EphA3 activation and ephrin-induced cytoskeletal remodelling. J. Cell Sci. 2016, 129, 277–289. [Google Scholar] [CrossRef]
- Espejo, R.; Jeng, Y.; Paulucci-Holthauzen, A.; Rengifo-Cam, W.; Honkus, K.; Anastasiadis, P.Z.; Sastry, S.K. PTP-PEST targets a novel tyrosine site in p120 catenin to control epithelial cell motility and Rho GTPase activity. J. Cell Sci. 2014, 127, 497–508. [Google Scholar] [CrossRef]
- Noren, N.K.; Liu, B.P.; Burridge, K.; Kreft, B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 2000, 150, 567–580. [Google Scholar] [CrossRef]
- Anastasiadis, P.Z.; Moon, S.Y.; Thoreson, M.A.; Mariner, D.J.; Crawford, H.C.; Zheng, Y.; Reynolds, A.B. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2000, 2, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Grosheva, I.; Shtutman, M.; Elbaum, M.; Bershadsky, A.D. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: A link between cell-cell contact formation and regulation of cell locomotion. J. Cell Sci. 2001, 114, 695–707. [Google Scholar] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Rhee, I. Important roles of protein tyrosine phosphatase PTPN12 in tumor progression. Pharm. Res. 2019, 144, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Chung, H.C.; Sun, T.; Tyagi, S.; Dobrolecki, L.E.; Dominguez-Vidana, R.; Kurley, S.J.; Orellana, M.; Renwick, A.; Henke, D.M.; et al. Combinatorial inhibition of PTPN12-regulated receptors leads to a broadly effective therapeutic strategy in triple-negative breast cancer. Nat. Med. 2018, 24, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Davidson, D.; Martins Souza, C.; Zhong, M.C.; Wu, N.; Park, M.; Muller, W.J.; Veillette, A. Loss of PTPN12 Stimulates Progression of ErbB2-Dependent Breast Cancer by Enhancing Cell Survival, Migration, and Epithelial-to-Mesenchymal Transition. Mol. Cell Biol. 2015, 35, 4069–4082. [Google Scholar] [CrossRef]
- Luo, R.Z.; Cai, P.Q.; Li, M.; Fu, J.; Zhang, Z.Y.; Chen, J.W.; Cao, Y.; Yun, J.P.; Xie, D.; Cai, M.Y. Decreased expression of PTPN12 correlates with tumor recurrence and poor survival of patients with hepatocellular carcinoma. PLoS ONE 2014, 9, e85592. [Google Scholar] [CrossRef]
- Lee, K.J.; Yoo, Y.H.; Kim, M.S.; Yadav, B.K.; Kim, Y.; Lim, D.; Hwangbo, C.; Moon, K.W.; Kim, D.; Jeoung, D.; et al. CD99 inhibits CD98-mediated beta1 integrin signaling through SHP2-mediated FAK dephosphorylation. Exp. Cell Res. 2015, 336, 211–222. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, S.H.; Yadav, B.K.; Ju, H.M.; Kim, M.S.; Park, J.H.; Jeoung, D.; Lee, H.; Hahn, J.H. The activation of CD99 inhibits cell-extracellular matrix adhesion by suppressing beta(1) integrin affinity. BMB Rep. 2012, 45, 159–164. [Google Scholar] [CrossRef]
- Guerzoni, C.; Fiori, V.; Terracciano, M.; Manara, M.C.; Moricoli, D.; Pasello, M.; Sciandra, M.; Nicoletti, G.; Gellini, M.; Dominici, S.; et al. CD99 triggering in Ewing sarcoma delivers a lethal signal through p53 pathway reactivation and cooperates with doxorubicin. Clin. Cancer Res. 2015, 21, 146–156. [Google Scholar] [CrossRef]
- Waclavicek, M.; Majdic, O.; Stulnig, T.; Berger, M.; Sunder-Plassmann, R.; Zlabinger, G.J.; Baumruker, T.; Stockl, J.; Ebner, C.; Knapp, W.; et al. CD99 engagement on human peripheral blood T cells results in TCR/CD3-dependent cellular activation and allows for Th1-restricted cytokine production. J. Immunol. 1998, 161, 4671–4678. [Google Scholar] [PubMed]
- Sohn, H.W.; Shin, Y.K.; Lee, I.S.; Bae, Y.M.; Suh, Y.H.; Kim, M.K.; Kim, T.J.; Jung, K.C.; Park, W.S.; Park, C.S.; et al. CD99 regulates the transport of MHC class I molecules from the Golgi complex to the cell surface. J. Immunol. 2001, 166, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.W.; Choi, E.Y.; Kim, S.H.; Lee, I.S.; Chung, D.H.; Sung, U.A.; Hwang, D.H.; Cho, S.S.; Jun, B.H.; Jang, J.J.; et al. Engagement of CD99 induces apoptosis through a calcineurin-independent pathway in Ewing’s sarcoma cells. Am. J. Pathol. 1998, 153, 1937–1945. [Google Scholar] [CrossRef]
- Lee, K.J.; Kim, Y.; Yoo, Y.H.; Kim, M.S.; Lee, S.H.; Kim, C.G.; Park, K.; Jeoung, D.; Lee, H.; Ko, I.Y.; et al. CD99-Derived Agonist Ligands Inhibit Fibronectin-Induced Activation of beta1 Integrin through the Protein Kinase A/SHP2/Extracellular Signal-Regulated Kinase/PTPN12/Focal Adhesion Kinase Signaling Pathway. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Zucchini, C.; Manara, M.C.; Pinca, R.S.; De Sanctis, P.; Guerzoni, C.; Sciandra, M.; Lollini, P.L.; Cenacchi, G.; Picci, P.; Valvassori, L.; et al. CD99 suppresses osteosarcoma cell migration through inhibition of ROCK2 activity. Oncogene 2014, 33, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.T.; Le Francois, B.G.; Goss, G.; Ding, K.; Bradbury, P.A.; Dimitroulakos, J. Lovastatin inhibits EGFR dimerization and AKT activation in squamous cell carcinoma cells: Potential regulation by targeting rho proteins. Oncogene 2010, 29, 4682–4692. [Google Scholar] [CrossRef]
- Mattila, P.K.; Batista, F.D.; Treanor, B. Dynamics of the actin cytoskeleton mediates receptor cross talk: An emerging concept in tuning receptor signaling. J. Cell Biol. 2016, 212, 267–280. [Google Scholar] [CrossRef]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct Mol. Biol. 2011, 18, 1244–1249. [Google Scholar] [CrossRef]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Zhai, J.; Lin, H.; Nie, Z.; Wu, J.; Canete-Soler, R.; Schlaepfer, W.W.; Schlaepfer, D.D. Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol Chem. 2003, 278, 24865–24873. [Google Scholar] [CrossRef]
- Westhoff, M.A.; Serrels, B.; Fincham, V.J.; Frame, M.C.; Carragher, N.O. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell Biol. 2004, 24, 8113–8133. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, S.; Takenawa, T. Regulation of cortical actin networks in cell migration. Int. Rev. Cytol. 2003, 229, 245–286. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Kurisu, S.; Takenawa, T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005, 96, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.Y.; Ohsugi, M.; Abe, Y.; Yamamoto, T. ZRP-1 controls Rho GTPase-mediated actin reorganization by localizing at cell-matrix and cell-cell adhesions. J. Cell Sci. 2007, 120, 2828–2837. [Google Scholar] [CrossRef]
- Fraguas, S.; Barberan, S.; Cebria, F. EGFR signaling regulates cell proliferation, differentiation and morphogenesis during planarian regeneration and homeostasis. Dev. Biol. 2011, 354, 87–101. [Google Scholar] [CrossRef]
- Song, Z.; Fusco, J.; Zimmerman, R.; Fischbach, S.; Chen, C.; Ricks, D.M.; Prasadan, K.; Shiota, C.; Xiao, X.; Gittes, G.K. Epidermal Growth Factor Receptor Signaling Regulates beta Cell Proliferation in Adult Mice. J. Biol. Chem. 2016, 291, 22630–22637. [Google Scholar] [CrossRef]
- Pennock, S.; Wang, Z. Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol. Cell Biol. 2003, 23, 5803–5815. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, C.; Croucher, D.R.; Soliman, M.A.; St-Denis, N.; Pasculescu, A.; Taylor, L.; Tate, S.A.; Hardy, W.R.; Colwill, K.; et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 2013, 499, 166–171. [Google Scholar] [CrossRef]
- Ayoub, E.; Hall, A.; Scott, A.M.; Chagnon, M.J.; Miquel, G.; Halle, M.; Noda, M.; Bikfalvi, A.; Tremblay, M.L. Regulation of the Src kinase-associated phosphoprotein 55 homologue by the protein tyrosine phosphatase PTP-PEST in the control of cell motility. J. Biol. Chem. 2013, 288, 25739–25748. [Google Scholar] [CrossRef]
- Trimble, W.S.; Grinstein, S. Barriers to the free diffusion of proteins and lipids in the plasma membrane. J. Cell Biol. 2015, 208, 259–271. [Google Scholar] [CrossRef]
- Jung, S.R.; Seo, J.B.; Shim, D.; Hille, B.; Koh, D.S. Actin cytoskeleton controls movement of intracellular organelles in pancreatic duct epithelial cells. Cell Calcium 2012, 51, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Manneville, J.B.; Etienne-Manneville, S.; Skehel, P.; Carter, T.; Ogden, D.; Ferenczi, M. Interaction of the actin cytoskeleton with microtubules regulates secretory organelle movement near the plasma membrane in human endothelial cells. J. Cell Sci. 2003, 116, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gross, D.J. Regulated EGF receptor binding to F-actin modulates receptor phosphorylation. Biochem. Biophys. Res. Commun. 2003, 312, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Den Hartigh, J.C.; van Bergen en Henegouwen, P.M.; Verkleij, A.J.; Boonstra, J. The EGF receptor is an actin-binding protein. J. Cell Biol. 1992, 119, 349–355. [Google Scholar] [CrossRef]
- Martinez-Munoz, L.; Rodriguez-Frade, J.M.; Barroso, R.; Sorzano, C.O.S.; Torreno-Pina, J.A.; Santiago, C.A.; Manzo, C.; Lucas, P.; Garcia-Cuesta, E.M.; Gutierrez, E.; et al. Separating Actin-Dependent Chemokine Receptor Nanoclustering from Dimerization Indicates a Role for Clustering in CXCR4 Signaling and Function. Mol. Cell 2018, 71, 873. [Google Scholar] [CrossRef]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef]
- Wehrle-Haller, B. Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 2012, 24, 569–581. [Google Scholar] [CrossRef]
- Li, S.Y.; Mruk, D.D.; Cheng, C.Y. Focal adhesion kinase is a regulator of F-actin dynamics: New insights from studies in the testis. Spermatogenesis 2013, 3, e25385. [Google Scholar] [CrossRef]
- Rae, J.M.; Scheys, J.O.; Clark, K.M.; Chadwick, R.B.; Kiefer, M.C.; Lippman, M.E. EGFR and EGFRvIII expression in primary breast cancer and cell lines. Breast Cancer Res. Treat. 2004, 87, 87–95. [Google Scholar] [CrossRef]
- Manara, M.C.; Terracciano, M.; Mancarella, C.; Sciandra, M.; Guerzoni, C.; Pasello, M.; Grilli, A.; Zini, N.; Picci, P.; Colombo, M.P.; et al. CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1R/RAS/Rac1 signaling. Oncotarget 2016, 7, 79925–79942. [Google Scholar] [CrossRef] [PubMed]
- Stock, J. Receptor signaling: Dimerization and beyond. Curr. Biol. 1996, 6, 825–827. [Google Scholar] [CrossRef]
- Weiss, A.; Schlessinger, J. Switching signals on or off by receptor dimerization. Cell 1998, 94, 277–280. [Google Scholar] [CrossRef]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Kusama, T.; Ralli, S.; Korc, M. Soluble type II transforming growth factor-beta (TGF-beta) receptor inhibits TGF-beta signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin. Cancer Res. 2001, 7, 2931–2940. [Google Scholar] [PubMed]
- Farooqi, A.A.; Siddik, Z.H. Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape. Cell Biochem. Funct. 2015, 33, 257–265. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Cao, S.; Kang, N.; Huebert, R.C.; Shah, V.H. Fibronectin induces endothelial cell migration through beta1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J. Biol Chem. 2012, 287, 7190–7202. [Google Scholar] [CrossRef]
- Chen, M.; She, H.; Kim, A.; Woodley, D.T.; Li, W. Nckbeta adapter regulates actin polymerization in NIH 3T3 fibroblasts in response to platelet-derived growth factor bb. Mol. Cell Biol. 2000, 20, 7867–7880. [Google Scholar] [CrossRef]
- Turk, H.F.; Chapkin, R.S. Analysis of epidermal growth factor receptor dimerization by BS(3) cross-linking. Methods Mol. Biol. 2015, 1233, 25–34. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-J.; Kim, Y.; Kim, M.S.; Ju, H.-M.; Choi, B.; Lee, H.; Jeoung, D.; Moon, K.-W.; Kang, D.; Choi, J.; et al. CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor. Cancers 2020, 12, 2895. https://doi.org/10.3390/cancers12102895
Lee K-J, Kim Y, Kim MS, Ju H-M, Choi B, Lee H, Jeoung D, Moon K-W, Kang D, Choi J, et al. CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor. Cancers. 2020; 12(10):2895. https://doi.org/10.3390/cancers12102895
Chicago/Turabian StyleLee, Kyoung-Jin, Yuri Kim, Min Seo Kim, Hyun-Mi Ju, Boyoung Choi, Hansoo Lee, Dooil Jeoung, Ki-Won Moon, Dongmin Kang, Jiwon Choi, and et al. 2020. "CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor" Cancers 12, no. 10: 2895. https://doi.org/10.3390/cancers12102895
APA StyleLee, K.-J., Kim, Y., Kim, M. S., Ju, H.-M., Choi, B., Lee, H., Jeoung, D., Moon, K.-W., Kang, D., Choi, J., Yook, J. I., & Hahn, J.-H. (2020). CD99–PTPN12 Axis Suppresses Actin Cytoskeleton-Mediated Dimerization of Epidermal Growth Factor Receptor. Cancers, 12(10), 2895. https://doi.org/10.3390/cancers12102895