Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms


Simple Summary
Abstract
1. Introduction
2. Telomerase Inhibition with Imetelstat in MF
3. Targeting Bone Marrow Fibrosis in MF
4. Activating p53: MDM2 Inhibition in PV and MF
5. Novel epigenetic therapies for MF
6. Targeting the Anti-Apoptotic Machinery in MF
7. Other Novel Targets in MF
8. Ropeginterferon alfa-2b in PV and Beyond
9. Givinostat for PV
10. Hepcidin Mimetics for Hematocrit Control in PV
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Maffioli, M. The role of JAK2 inhibitors in MPNs 7 years after approval. Blood 2018, 131, 2426–2435. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, M.M.; Jones, K.; Sun, M.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef]
- Kuykendall, A.T.; Shah, S.; Talati, C.; Al Ali, N.; Sweet, K.; Padron, E.; Sallman, D.A.; Lancet, J.E.; List, A.F.; Zuckerman, K.S.; et al. Between a rux and a hard place: Evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann. Hematol. 2018, 97, 435–441. [Google Scholar] [CrossRef]
- Palandri, F.; Breccia, M.; Bonifacio, M.; Polverelli, N.; Elli, E.M.; Benevolo, G.; Tiribelli, M.; Abruzzese, E.; Lurlo, A.; Heidel, F.H.; et al. Life after ruxolitinib: Reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer 2020, 126, 1243–1252. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naive patients with myelofibrosis. J. Clin. Oncol. 2017, 35, 3844. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2017, 5, 73–81. [Google Scholar] [CrossRef]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): An international, randomised, phase 3 trial. Lancet Haematol. 2017, 4, 225–236. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T.; Savona, M.R.; Scott, B.L.; Talpaz, M.; Egyed, M.; Harrison, C.N.; Yacoub, A.; Vannucchi, A.M.; Mead, A.J.; O’Sullivan, J.; et al. Results of PAC203: A randomized phase 2 dose-finding study and determination of the recommended dose of pacritinib in patients with myelofibrosis. Blood 2019, 134, 667. [Google Scholar] [CrossRef]
- Kuykendall, A.; Horvat, N.P.; Pandey, G.; Komrokji, R.S.; Reuther, G.W. Finding a jill for JAK: Assessing past, present and future JAK inhibitor combination approaches in myelofibrosis. Cancers Basel 2020, 12, 2278. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.O.; Kosiorek, H.E.; Prchal, J.T.; Rambaldi, A.; Berenzon, D.; Yacoub, A.; Harrison, C.N.; McMullin, M.F.; Vannucchi, A.M.; Ewing, J.; et al. Results of the myeloproliferative neoplasms-research consortium (MPN-RC) 112 randomized trial of pegylated interferon alfa-2a (PEG) versus hydroxyurea (HU) therapy for the treatment of high risk polycythemia vera (PV) and high risk essential thrombocythemia (ET). Blood 2018, 132, 577. [Google Scholar]
- European Medicines Agency. Besremi. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/besremi (accessed on 30 August 2019).
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet. Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Masciulli, A.; Ferrari, A.; Carobbio, A.; Ghirardi, A.; Barbui, T. Ruxolitinib for the prevention of thrombosis in polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2020, 4, 380–386. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Zachee, P.; Hino, M.; Pane, F.; Masszi, T.; Harrison, C.N.; Mesa, R.; Miller, C.B.; Passamonti, F.; Durrant, S.; et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 2020, 7, 184–185. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; Finazzi, G.; Finazzi, M.C.; Masciulli, A.; Carobbio, A.; Ghirardi, A.; Tognoni, G. A reappraisal of the benefit-risk profile of hydroxyurea in polycythemia vera: A propensity-matched study. Am. J. Hematol. 2017, 92, 1131–1136. [Google Scholar] [CrossRef]
- De Stefano, V.; Rossi, E.; Carobbio, A.; Ghirardi, A.; Betti, S.; Finazzi, G.; Vannucchi, A.M.; Barbui, T. Hydroxyurea prevents arterial and late venous thrombotic recurrences in patients with myeloproliferative neoplasms but fails in the splanchnic venous district. Pooled analysis of 1500 cases. Blood Cancer J. 2018, 8, 112. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pungolino, E.; Malabarba, L.; Bertazzoni, P.; Valentini, M.; Orlandi, E.; Arcaini, L.; Brusamolino, E.; Pascutto, C.; et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am. J. Med. 2004, 117, 755–761. [Google Scholar] [CrossRef]
- Carobbio, A.; Antonioli, E.; Guglielmelli, P.; Vannucchi, A.M.; Delaini, F.; Guerini, V.; Finazzi, G.; Rambaldi, A.; Barbui, T. Leukocytosis and risk stratification assessment in essential thrombocythemia. J. Clin. Oncol. 2008, 26, 2732–2736. [Google Scholar] [CrossRef]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.J.; Forsyth, C.; Harrison, C.N.; Green, A.R. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Gotic, M.; Holowiecki, J.; Penka, M.; Thiele, J.; Kvasnicka, H.M.; Kralovics, R.; Petrides, P.E. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: The ANAHYDRET Study, a randomized controlled trial. Blood 2013, 121, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Essential thrombocythemia. N. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef]
- Harrison, C.N.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017, 130, 1889–1897. [Google Scholar] [CrossRef]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rumi, E.; Gattoni, E.; Pieri, L.; Zhen, H.; Granier, M.; Assad, A.; et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: Long-term phase 2 study results. Blood 2017, 130, 1768–1771. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef]
- Wang, X.; Hu, C.S.; Petersen, B.; Qiu, J.; Ye, F.; Houldsworth, J.; Eng, K.; Huang, F.; Hoffman, R. Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells. Blood Adv. 2018, 2, 2378–2388. [Google Scholar] [CrossRef]
- Mascarenhas, J.O.; Komrokji, R.S.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Martino, B.; Hoffman, R.; Odenike, O.; Bussolari, J.; et al. Favorable overall survival with imetelstat treatment correlates with other clinical benefits in intermediate 2 or high risk myelofibrosis relapsed/refractory to janus kinase inhibitor. Hemasphere 2020, 4, 1107. [Google Scholar]
- Mascarenhas, J.O.; Komrokji, R.S.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Telomerase activity, telomere length and htert expression correlate with clinical outcomes in higher risk myelofibrosis (mf) relapsed/refractory (r/r) to janus kinase inhibitor treated with imetelstat. Hemasphere 2020, 4, 1098. [Google Scholar]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.; Mascarenhas, J.O.; Komrokji, R.S.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; et al. Imetelstat treatment results in clinical benefits, including improved overall survival, in patients with higher-risk triple-negative myelofibrosis relapsed/refractory to janus kinase inhibitors (jaki). Hemasphere 2020, 4, 1101. [Google Scholar]
- Baerlocher, G.M.; Oppliger, E.L.; Ottmann, O.G.; Spitzer, G.; Odenike, O.; McDevitt, M.A.; Röth, A.; Daskalakis, M.; Burington, B.; Stuart, M.; et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N. Engl. J. Med. 2015, 373, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Osawa, Y.; Izumi, T.; Nagao, S.; Takano, K.; Okada, Y.; Tachi, N.; Teramoto, M.; Kawamura, T.; Horiuchi, T.; et al. Myeloproliferative leukemia protein activation directly induces fibrocyte differentiation to cause myelofibrosis. Leukemia 2017, 31, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Kato, S.; Kawamura, T.; Takada, K.; Sone, T.; Ogata, H.; Saito, K.; Izumi, T.; Nagao, S.; Takano, K.; et al. Increased SLAMF7(high) monocytes in myelofibrosis patients harboring JAK2V617F provide a therapeutic target of elotuzumab. Blood 2019, 134, 814–825. [Google Scholar] [CrossRef]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell 2017, 20, 785–800. [Google Scholar] [CrossRef]
- Agarwal, A.; Morrone, K.; Bartenstein, M.; Zhao, Z.J.; Verma, A.; Goel, S. Bone marrow fibrosis in primary myelofibrosis: Pathogenic mechanisms and the role of TGF-beta. Stem Cell Investig. 2016, 3, 5. [Google Scholar]
- Ozono, Y.; Shide, K.; Kameda, T.; Kamiunten, A.; Tahira, Y.; Sekine, M.; Akizuki, K.; Nakamura, K.; Iwakiri, H.; Sueta, M.; et al. Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Lupher, M.L., Jr. PRM-151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect. 2010, 23, 305–315. [Google Scholar] [CrossRef]
- Verstovsek, S.; Hasserjian, R.P.; Pozdnyakova, O.; Veletic, I.; Mesa, R.A.; Foltz, L.; Mascarenhas, J.; Ritchie, E.K.; Palmer, J.; Silver, R.T.; et al. PRM-151 in myelofibrosis: Efficacy and safety in an open label extension study. Blood 2018, 132, 686. [Google Scholar] [CrossRef]
- Verstovsek, S.; Talpaz, M.; Wadleigh, M.; Palmer, J.; Isidori, A.; te Boekhorst, P.A.; Savona, M.R.; Gotlib, J.R.; Hasserjian, R.P.; Pozdnyakova, O.; et al. A randomized, double blind phase 2 study of 3 different doses of prm-151 in patients with myelofibrosis who were previously treated with or ineligible for ruxolitinib. Hemasphere 2019, 3, 828. [Google Scholar] [CrossRef]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Dong, H.; Maltzman, J.D.; Sharma, S.; Silverman, J.; Oh, S.T.; Gotlib, J. A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Br. J. Haematol. 2017, 176, 939–949. [Google Scholar] [CrossRef]
- Wen, Q.J.; Yang, Q.; Goldenson, B.; Malinge, S.; Lasho, T.; Schneider, R.K.; Breyfogle, L.J.; Schultz, R.; Gilles, L.; Koppikar, P.; et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat. Med. 2015, 21, 1473–1480. [Google Scholar] [PubMed]
- Gangat, N.; Marinaccio, C.; Swords, R.; Watts, J.M.; Gurbuxani, S.; Rademaker, A.; Fought, A.J.; Frankfurt, O.; Altman, J.K.; Wen, Q.J.; et al. Aurora Kinase a inhibition provides clinical benefit, normalizes megakaryocytes, and reduces bone marrow fibrosis in patients with myelofibrosis. Clin. Cancer Res. 2019, 25, 4898–4906. [Google Scholar] [CrossRef]
- Tefferi, A.; Cervantes, F.; Mesa, R.; Passamonti, F.; Verstovsek, S.; Vannucchi, A.M.; Gotlib, J.; Dupriez, B.; Pardanani, A.; Harrison, C.; et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013, 122, 1395–1398. [Google Scholar] [CrossRef]
- Gilles, L.; Arslan, A.D.; Marinaccio, C.; Wen, Q.J.; Arya, P.; McNulty, M.; Yang, Q.; Zhao, J.C.; Konstantinoff, K.; Lasho, T.; et al. Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J. Clin. Investig. 2017, 127, 1316–1320. [Google Scholar] [CrossRef]
- Zingariello, M.; Sancillo, L.; Martelli, F.; Ciaffoni, F.; Marra, M.; Varricchio, L.; Rana, R.A.; Zhao, C.; Crispino, J.D.; Migliaccio, A.R.; et al. The thrombopoietin/MPL axis is activated in the Gata1(low) mouse model of myelofibrosis and is associated with a defective RPS14 signature. Blood Cancer J. 2017, 7, e572. [Google Scholar] [CrossRef]
- Zingariello, M.; Martelli, F.; Ciaffoni, F.; Masiello, F.; Ghinassi, B.; D’Amore, E.; Massa, M.; Barosi, G.; Sancillo, L.; Li, X.; et al. Characterization of the TGF-beta1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood 2013, 121, 3345–3363. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. P53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef]
- Nakatake, M.; Monte-Mor, B.; Debili, N.; Casadevall, N.; Ribrag, V.; Solary, E.; Vainchenker, W. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via la expression in myeloproliferative neoplasms. Oncogene 2012, 31, 1323–1333. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Li, Y.; Wang, X.; Hoffman, R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon alpha 2a target JAK2V617F-positive progenitor and stem cells. Blood 2014, 124, 771–779. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Lu, M.; Kosiorek, H.; Virtgaym, E.; Xia, L.; Sandy, L.; Mesa, R.; Petersen, B.; Farnoud, N.; Najfeld, V.; et al. Oral idasanutlin in patients with polycythemia vera. Blood 2019, 134, 525–533. [Google Scholar] [CrossRef]
- Barosi, G.; Mesa, R.; Finazzi, G.; Harrison, C.; Kiladjian, J.J.; Lengfelder, E.; McMullin, M.F.; Passamonti, F.; Vannucchi, A.M.; Besses, C.; et al. Revised response criteria for polycythemia vera and essential thrombocythemia: An ELN and IWG-MRT consensus project. Blood 2013, 121, 4778–4781. [Google Scholar] [CrossRef]
- Al-Ali, H.; Delgado, R.G.; Lange, A.; Pluta, A.; McLornan, D.P.; Vachhani, P.; Damaj, G.; Jost, P.J.; Rejto, L.; Hus, M.; et al. KRT-232, A first-in-class, murine double minute 2 inhibitor, for myelofibrosis relapsed or refractory to janus-associated kinase inhibitor treatment. Hemasphere 2020, 4, 215. [Google Scholar]
- Quintas-Cardama, A.; Tong, W.; Kantarjian, H.; Thomas, D.; Ravandi, F.; Kornblau, S.; Manshouri, T.; Cortes, J.E.; Garcia-Manero, G.; Verstovsek, S. A phase II study of 5-azacitidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia 2008, 22, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Estrov, Z.; Borthakur, G.; Cortes, J.; Verstovsek, S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012, 36, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Lu, M.; Li, T.; Petersen, B.; Hochman, T.; Najfeld, V.; Goldberg, J.D.; Hoffman, R. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br. J. Haematol. 2013, 161, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Sandy, L.; Lu, M.; Yoon, J.; Petersen, B.; Zhang, D.; Ye, F.; Newsom, C.; Najfeld, V.; Hochman, T.; et al. A phase II study of panobinostat in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk. Res. 2016, 53, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; de Groote, S.; Papalexi, E.; Somasundara, A.V.H.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Harrison, C.N.; Patriarca, A.; Devos, T.; Palandri, F.; Rampal, R.K.; Mead, A.; Kremyanskaya, M.; Somervaille, T.; Wondergem, M.; et al. CPI-0610, a bromodomain and extraterminal domain protein (bet) inhibitor, in combination with ruxolitinib, in jak inhibitor treatment naïve myelofibrosis patients: Update from manifest phase 2 study. Haemasphere 2020, 4, 1084. [Google Scholar]
- Verstovsek, S.; Mascarenhas, J.; Kremyanskaya, M.; Hoffman, R.; Rampal, R.K.; Gupta, V.; Talpaz, M.; Granacher, N.; Leber, B.; Kiladjian, J.-J.; et al. CPI-0610, bromodomain and extraterminal domain protein (bet) inhibitor, as ’add-on’ to ruxolitinib (rux), in advanced myelofibrosis patients with suboptimal response: Update of manifest phase 2 study. Haemasphere 2020, 4, 1083. [Google Scholar]
- Talpaz, M.; Rampal, R.K.; Verstovsek, S.; Harrison, C.N.; Drummond, M.W.; Kiladjian, J.; Vannucchi, A.M.; Kremyanskaya, M.; Schiller, G.J.; Patriarca, A.; et al. CPI-0610, a bromodomain and extraterminal domain protein (BET) inhibitor, as monotherapy in advanced myelofibrosis patients refractory/intolerant to JAK inhibitor: Update from phase 2 MANIFEST study. Hemasphere 2020, 4, 1091. [Google Scholar]
- Kleppe, M.; Shank, K.; Efthymia, P.; Riehnhoff, H.; Levine, R.L. Lysine-specific histone demethylase, LSD1, (KDM1A) as a novel therapeutic target in myeloproliferative neoplasms. Blood 2015, 126, 601. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y., Jr.; Levine, R.L.; et al. LSD1 Inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere 2018, 2, 54. [Google Scholar] [CrossRef] [PubMed]
- Pettit, K.; Yacoub, A.; Gerds, A.T.; Bradley, T.J.; Tatarczuch, M.; Curtin, N.J.; Shortt, J.; Rossetti, J.M.; Burbury, K.; Ewing, J.C.; et al. A phase 2 study of bomedemstat (IMG-7289), a lysine-specific demethylase-1 (LSD1) inhibitor, for the treatment of myelofibrosis (MF). Hemasphere 2020, 4, 1080. [Google Scholar]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 inhibition modulates E2F1 methylation and gene regulatory networks leading to therapeutic efficacy in JAK2V617F mutant MPN. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.S.; Bracken, L.S.; et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Garcia, J.S.; Mesa, R.A.; Somervaille, T.; Ritchie, E.K.; Komrokji, R.S.; Pemmaraju, N.; Jamieson, C.; Papadantonakis, N.; Foran, J.M.; et al. Navitoclax in combination with ruxolitinib in patients with primary or secondary myelofibrosis: A phase 2 study. Haemasphere 2020, 4, 1081. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.S.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase 2 study of single agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 8. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Packham, G.; White, E.L.; Eischen, C.M.; Yang, H.; Parganas, E.; Ihle, J.N.; Grillot, D.A.M.; Zambetti, G.P.; Nuñez, G.; Cleveland, J.L. Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev. 1998, 12, 2475–2487. [Google Scholar] [CrossRef]
- Petiti, J.; Lo Iacono, M.; Rosso, V.; Andreani, G.; Jovanovski, A.; Podesta, M.; Lame, D.; de Gobbi, M.; Fava, C.; Saglio, G.; et al. Bcl-xL represents a therapeutic target in Philadelphia negative myeloproliferative neoplasms. J. Cell Mol. Med. 2020, 24, 10978–10986. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Heaton, W.L.; Senina, A.V.; Pomicter, A.D.; Salama, M.E.; Clair, P.M.; Yan, D.; Bell, R.N.; Gililland, J.M.; Prchal, J.T.; O’Hare, T.; et al. Autocrine Tnf signaling favors malignant cells in myelofibrosis in a Tnfr2-dependent fashion. Leukemia 2018, 32, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Carter, B.Z.; Kantarjian, H.M.; Cortes, J.E.; Bose, P.; Kadia, T.M.; Garcia-Manero, G.; Bueso-Ramos, C.E.; DiNardo, C.D.; Bledsoe, S.; et al. Final Results of Phase 2 Clinical Trial of LCL161, a Novel Oral SMAC Mimetic/IAP Antagonist for Patients with Intermediate to High Risk Myelofibrosis. Blood 2019, 134, 555. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Gupta, V.; Ali, H.; Yacoub, A.; Wang, E.S.; Lee, S.; Schiller, G.; Hoberman, M.; Wysowskyj, H.; Chen, J.; et al. Updated results from a phase 1/2 clinical trial of tagraxofusp, a CD123-targeted therapy, in patients with poor-risk myelofibrosis. Hemasphere 2020, 4, 218. [Google Scholar]
- Yan, D.; Pomicter, A.D.; Tantravahi, S.; Mason, C.C.; Senina, A.V.; Ahmann, J.M.; Wang, Q.; Than, H.; Patel, A.B.; Heaton, W.L.; et al. Nuclear-cytoplasmic transport is a therapeutic target in myelofibrosis. Clin. Cancer Res. 2018, 25, 14. [Google Scholar] [CrossRef]
- Wang, J.C.; Chen, C.; Kundra, A.; Kodali, S.; Pandey, A.; Wong, C.; Cheung, T.; Gotlieb, V.; Joseph, G.; Tribie, S. Programmed cell death receptor (PD-1) ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk. Res. 2019, 79, 52–59. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef]
- Gisslinger, H.; Zagrijtschuk, O.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Strecker, K.; et al. Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015, 126, 1762–1769. [Google Scholar] [CrossRef]
- Gisslinger, H.; Buxhofer-Ausch, V.; Thaler, J.; Forjan, E.; Willenbacher, E.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Stampfl-Mattersberger, M.; Egle, A.; et al. Long-Term efficacy and safety of ropeginterferon alfa-2b in patients with polycythemia vera—Final phase I/II peginvera study results. Blood 2018, 132, 3030. [Google Scholar] [CrossRef]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, 196–208. [Google Scholar] [CrossRef]
- Kiladjian, J.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illés, Á.; Pylypenko, H.; et al. Thromboembolic risk reduction and high rate of complete molecular response with long-term use of ropeginterferon alpha-2b in polycythemia vera: Results from a randomized controlled study. Blood 2019, 134, 553. [Google Scholar] [CrossRef]
- Gisslinger, H.; Gisslinger, B.; Schalling, M.; Krejcy, K.; Widmann, R.S.; Kralovics, R.; Krauth, M.-T. Effect of ropeginterferon alfa-2b in prefibrotic primary myelofibrosis. Blood 2018, 132, 3029. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; De Stefano, V.; Masciulli, A.; Carobbio, A.; Ghirardi, A.; Ciceri, F.; Bonifacio, M.; Iurlo, A.; Palandri, F.; et al. Phase II randomized clinical trial comparing ropeginterferon versus phlebotomy in low-risk patients with polycythemia vera. results of the pre-planned interim analysis. Hemasphere 2020, 4, 2602. [Google Scholar]
- Bose, P.; Verstovsek, S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2016, 25, 1393–1403. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Bose, P.; Verstovsek, S. Givinostat: An emerging treatment for polycythemia vera. Expert Opin. Investig. Drugs 2020, 29, 525–536. [Google Scholar] [CrossRef]
- Guerini, V.; Barbui, V.; Spinelli, O.; Salvi, A.; Dellacasa, C.; Carobbio, A.; Introna, M.; Barbui, T.; Golay, J.; Rambaldi, A. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia 2008, 22, 740–747. [Google Scholar] [CrossRef]
- Calzada, A.A.; Pedrini, O.; Finazzi, G.; Leoni, F.; Mascagni, P.; Introna, M.; Rambaldi, A.; Golay, J. Associazione Italiana per la Ricerca sul Cancro-Gruppo Italiano Malattie Mieloproliferative Investigators. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2(V617F) myeloproliferative neoplasm patients. Exp. Hematol. 2013, 41, 253. [Google Scholar] [CrossRef]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef]
- Rambaldi, A.; Iurlo, A.; Vannucchi, A.M.; Noble, R.; von Bubnoff, N.; Guarini, A.; Martino, B.; Pezzutto, A.; Carli, G.; De Muro, M.; et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: A two-part Phase Ib/II study. Leukemia 2020, 34, 2234–2237. [Google Scholar] [CrossRef]
- Ginzburg, Y.Z.; Feola, M.; Zimran, E.; Varkonyi, J.; Ganz, T.; Hoffman, R. Dysregulated iron metabolism in polycythemia vera: Etiology and consequences. Leukemia 2018, 32, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T.; Vannucchi, A.M.; Passamonti, F.; Kremyanskaya, M.; Gotlib, J.R.; Palmer, J.; McCaul, K.; Ribrag, V.; Mead, A.J.; Harrison, C.N.; et al. A phase 2 study of luspatercept in patients with myelofibrosis-associated anemia. Blood 2019, 134, 557. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J. Clin. Oncol. 2017, 36, 15. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef]
- Green, A.; Beer, P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N. Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef]
- Patel, A.A.; Cahill, K.; Charnot-Katsikas, A.; Liu, H.; Gurbuxani, S.; Thirman, M.; Kosuri, S.; Artz, A.S.; Larson, R.A.; Stock, W.; et al. Clinical outcomes of IDH2-mutated advanced-phase Ph-negative myeloproliferative neoplasms treated with enasidenib. Br. J. Haematol. 2020, 190, 48–51. [Google Scholar] [CrossRef]



{kind=link}
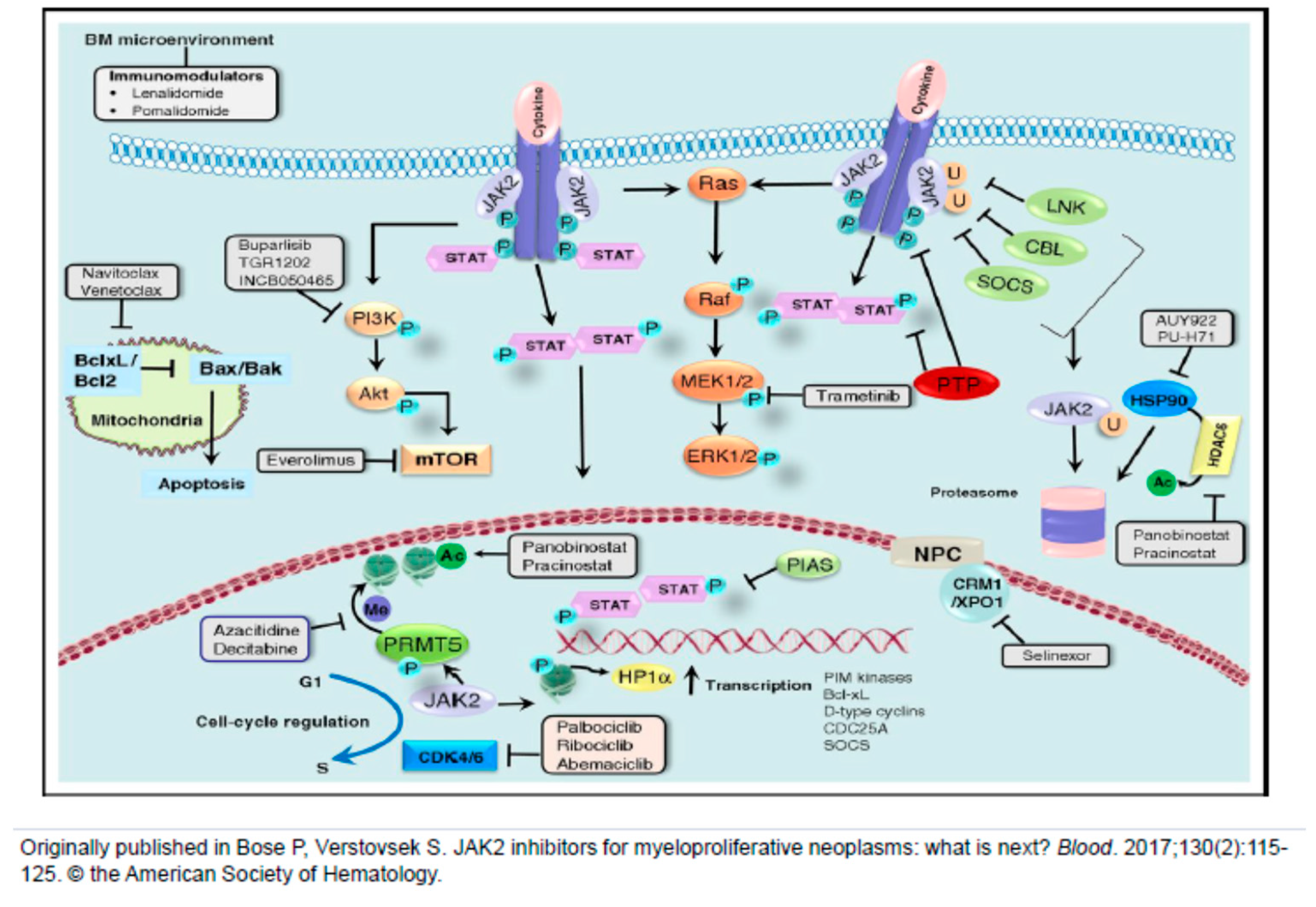{kind=link}
| Documented Progressive Disease during or after JAK Inhibitor Therapy: |
|---|
|
| RELAPSED: Progressive disease any time while on ruxolitinib/JAK inhibitor |
| Defined as: Increase in spleen volume by ≥25% from nadir by MRI/CT Appearance of new splenomegaly palpable ≥5 cm below LCM ≥100% increase in palpable distance below LCM for baseline splenomegaly of 5–10 cm ≥50% increase in palpable distance below LCM for baseline splenomegaly of >10 cm |
| REFRACTORY:Lack of spleen response after ≥ 12 weeks of ruxolitinib/JAK inhibitor |
| Defined as: Persistent splenomegaly, by physical exam, that is palpable ≥ 5 cm below the left LCM AND TSS of ≥ 10 by MPN-SAF TSS 2.0 or single symptom score ≥ 5 or two symptom scores ≥ 3, including only the symptoms of LU quadrant pain, bone pain, itching or night sweats |
| Agent | MOA/Drug Class (Route) | Clinicaltrials.Gov Identifier | Phase | Primary Objective or Endpoint | Results [ref.] |
|---|---|---|---|---|---|
| Imetelstat [40] | Telomerase inhibitor (IV) | NCT02426086 | 2 (n = 107) | ≥35% SVR and ≥50% TSS reduction rates at 24 weeks; OS a key secondary endpoint | Spleen and symptom response rates at 24 weeks 10.2% (six of 59) and 32.2% (19 of 59); median OS 28.1 months (all 9.4 mg/kg arm) |
| PRM-151 [54] | Anti-fibrotic agent (IV) | NCT01981850 | 2 (n = 97) | BM fibrosis reduction by ≥1 grade at any time | 27.8% (27 of 97) had BM fibrosis grade decrease at any time; 34% (32/94) had ≥50% TSS reduction; 41% (31/76) had some SVR |
| CPI-0610 (monotherapy) [78] | BET inhibitor (oral) | NCT02158858 | 2 (n = 43) | ≥35% SVR rate in non-RBC TD patients; rate of TI in RBC TD patients | TD→TI conversion rate 21% (3/14 evaluable) and ≥35% SVR rate 24% (5/21 evaluable) at 24 weeks |
| Bomedemstat [81] | LSD1 inhibitor (oral) | NCT03136185 | 2 (n = 36) | Safety, PK, SVR | Most common TEAE dysgeusia (33%); no DLTs; some SVR in 83% (10/12) and TSS reduction in 86% (12/14) of evaluable pts |
| KRT-232 [70] | MDM2 inhibitor (oral) | NCT03662126 | 2 (n = 82) | ≥35% SVR at week 24 | Best spleen response rate 16% (four of 25 evaluable) and best symptom response rate 30% (eight of 27 evaluable) at 240 mg on d1-7 q28d |
| LCL-161 [95] | Smac-mimetic (oral) | NCT02098161 | 2 (n = 50) | ORR by IWG-MRT 2013 criteria | 30% ORR (15 of 50 pts); six anemia responders |
| Tagraxofusp [96] | CD123-directed fusion protein (IV) | NCT02268253 | 1/2 (n = 32) | Determination of RP2D; defining efficacy and safety | CLS occurred in three pts (9%); IWG-MRT 2013 spleen response in one pt; 56% (10 of 18 evaluable) had some spleen size reduction; 46% (11 of 24 evaluable) had some symptom improvement |
| Alisertib [57] | Aurora kinase inhibitor (oral) | NCT02530619 | Pilot (n = 24) | Safety; response assessment by IWG-MRT 2013 criteria | Most frequent TEAEs cytopenias, N/V/D, mucositis; response rates in evaluable pts: spleen 29% (4/14), symptoms 32% (7/22), anemia 11% (2/19) |
| Luspatercept (monotherapy) [100] | Activin receptor ligand trap (subcutaneous) | NCT03194542 | 2 (n = 43) | Anemia response: Hgb improvement or RBC TI | 14% (n = 22) had Hgb improvement and 10% (n = 21) achieved RBC TI (with monotherapy) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bose, P.; Masarova, L.; Verstovsek, S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers 2020, 12, 2891. https://doi.org/10.3390/cancers12102891
Bose P, Masarova L, Verstovsek S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers. 2020; 12(10):2891. https://doi.org/10.3390/cancers12102891
Chicago/Turabian StyleBose, Prithviraj, Lucia Masarova, and Srdan Verstovsek. 2020. "Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms" Cancers 12, no. 10: 2891. https://doi.org/10.3390/cancers12102891
APA StyleBose, P., Masarova, L., & Verstovsek, S. (2020). Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers, 12(10), 2891. https://doi.org/10.3390/cancers12102891