Exploiting Chromosomal Instability of PTEN-Deficient Triple-Negative Breast Cancer Cell Lines for the Sensitization Against PARP1 Inhibition in a Replication-Dependent Manner

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
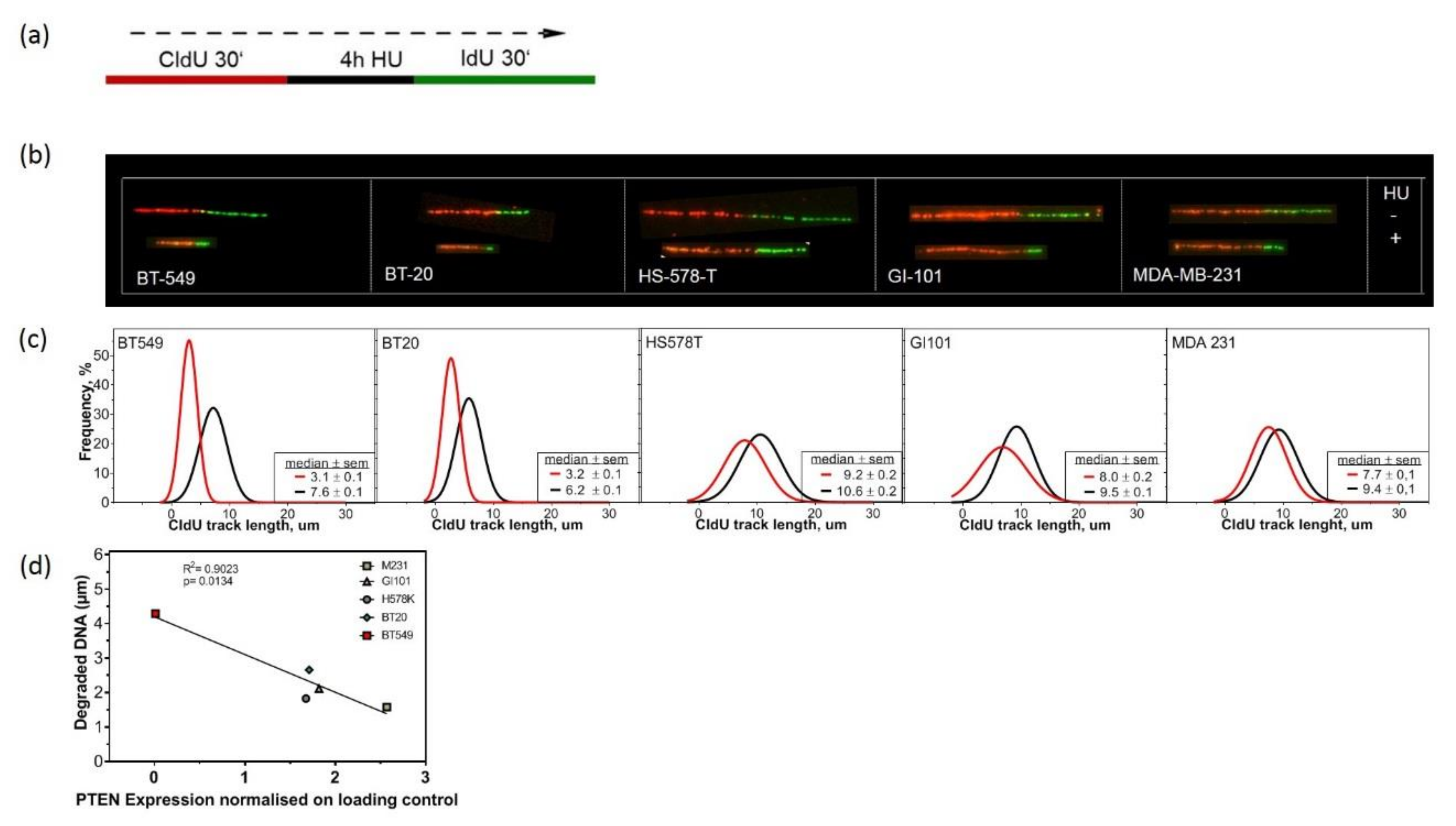
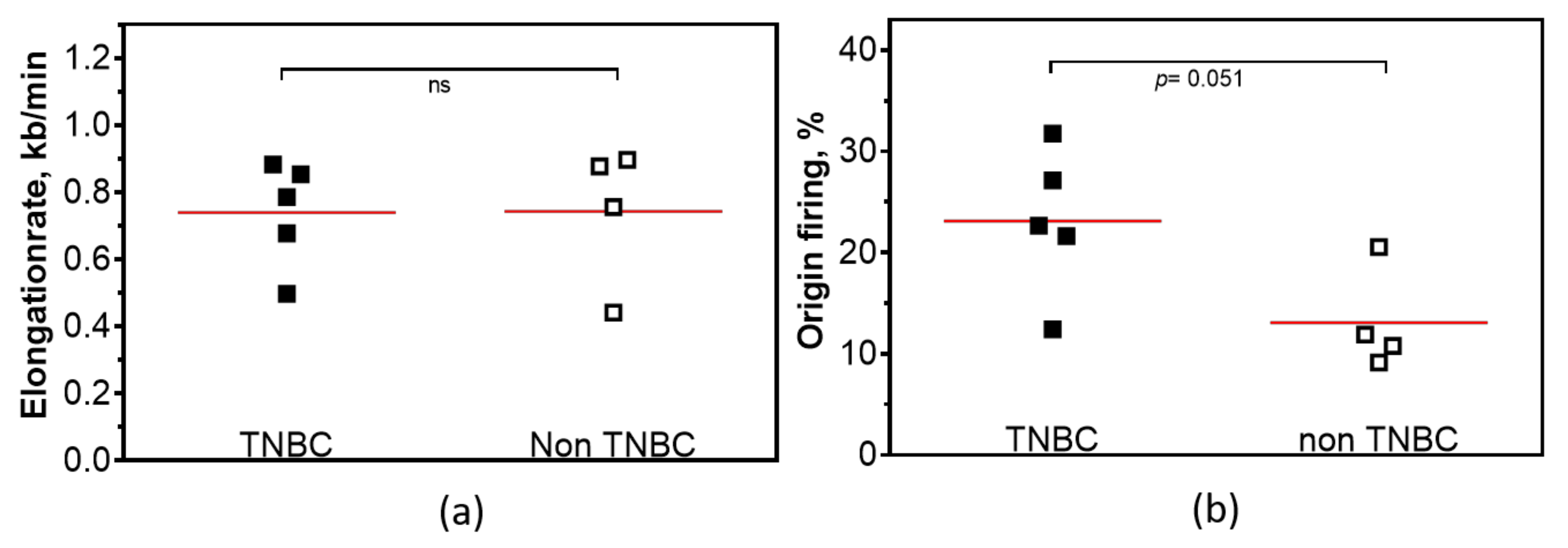
2.1. Low PTEN Expression Levels Correlate with CIN
2.2. Low Expression of PTEN Causes Replication Stress in TNBC Cell Lines
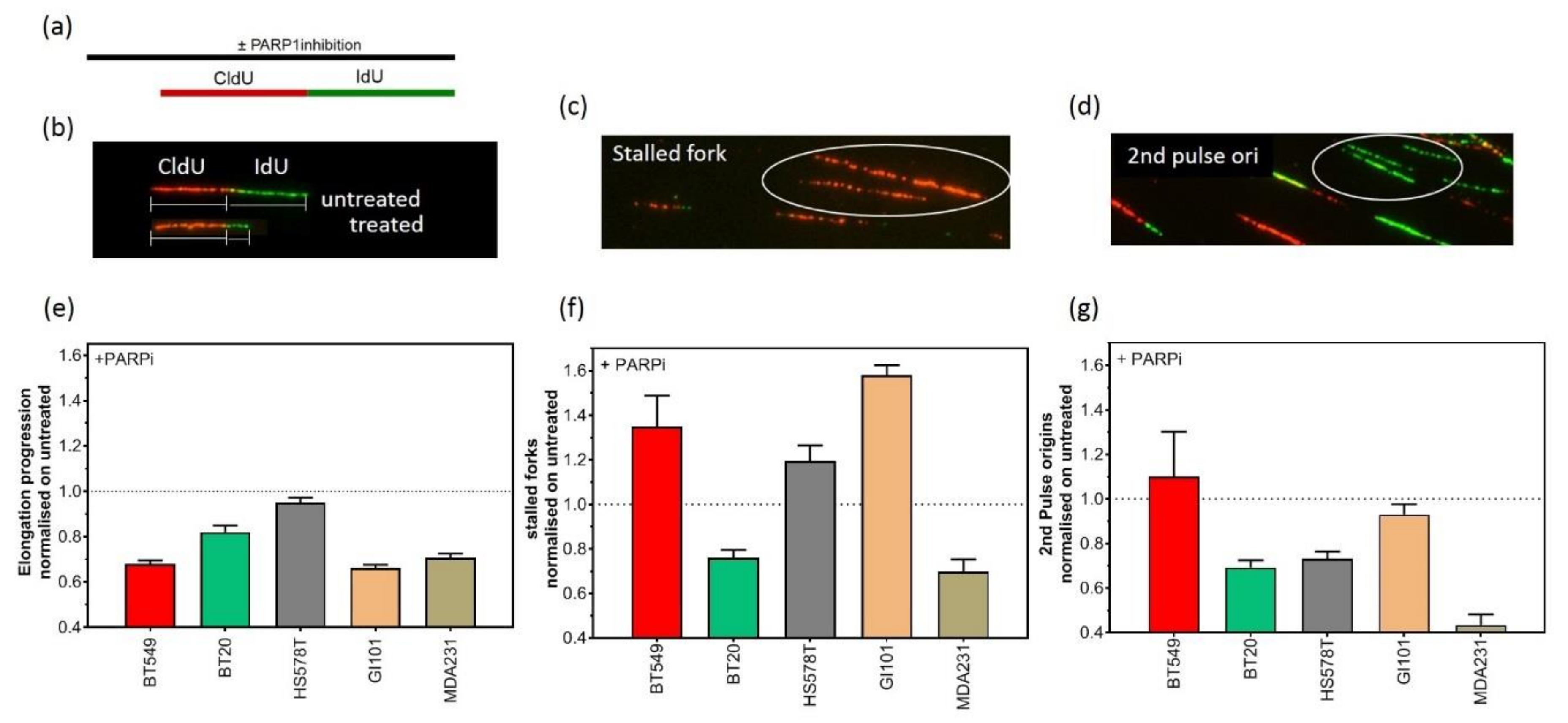
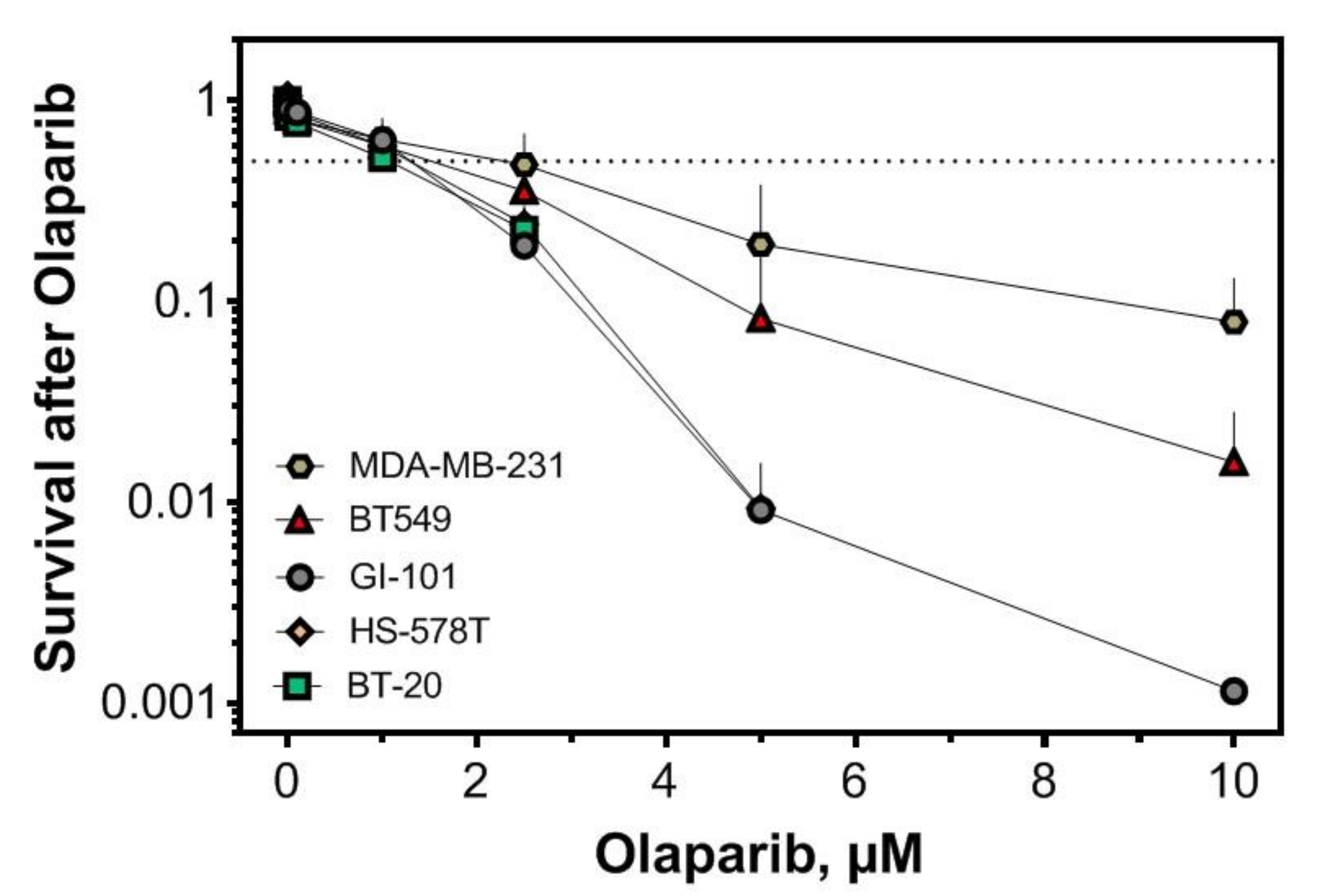
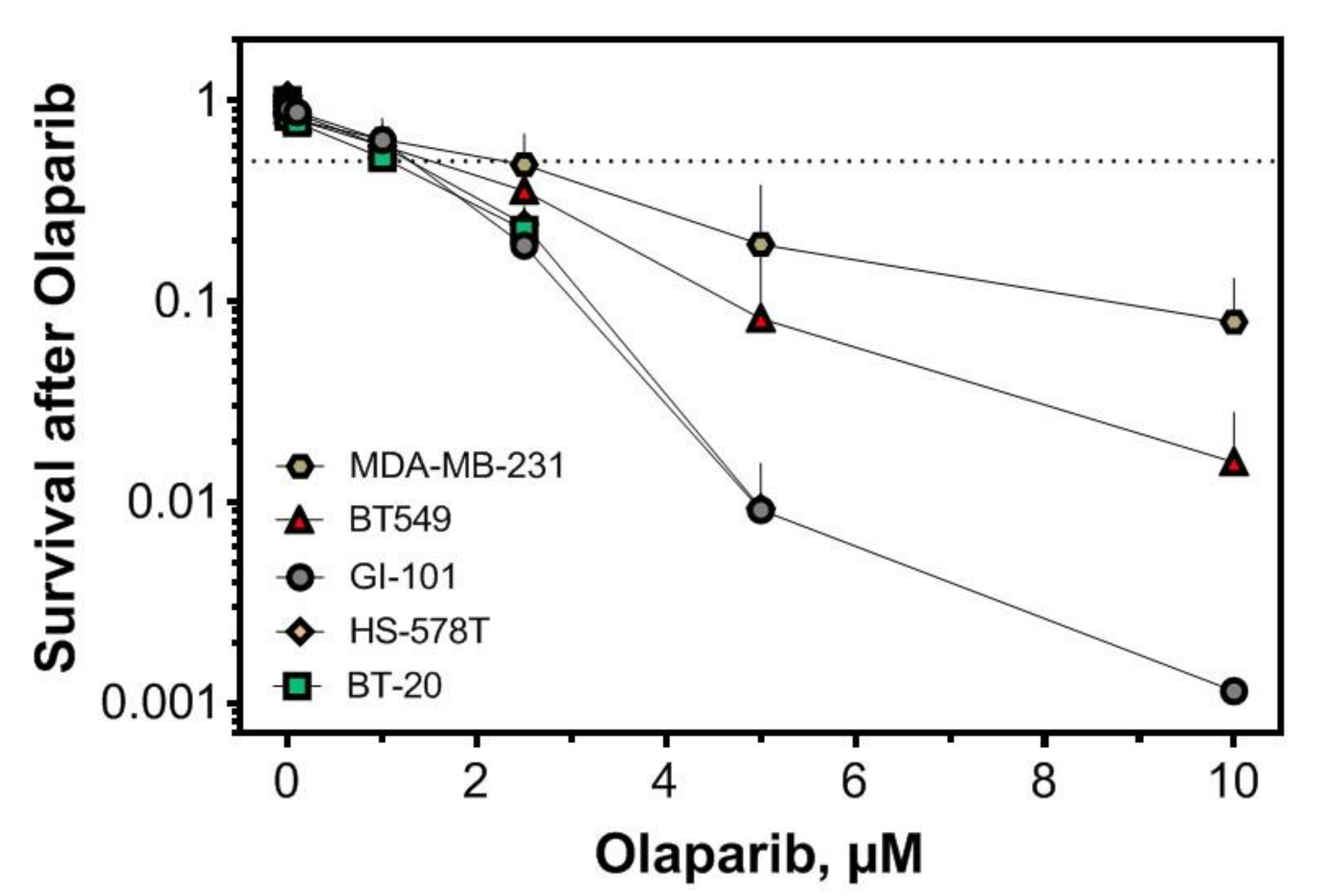
2.3. Replication-Dependent Sensitization by PARP1 Inhibition Due to Reduced Replication Fork Elongation and Fork Stalling
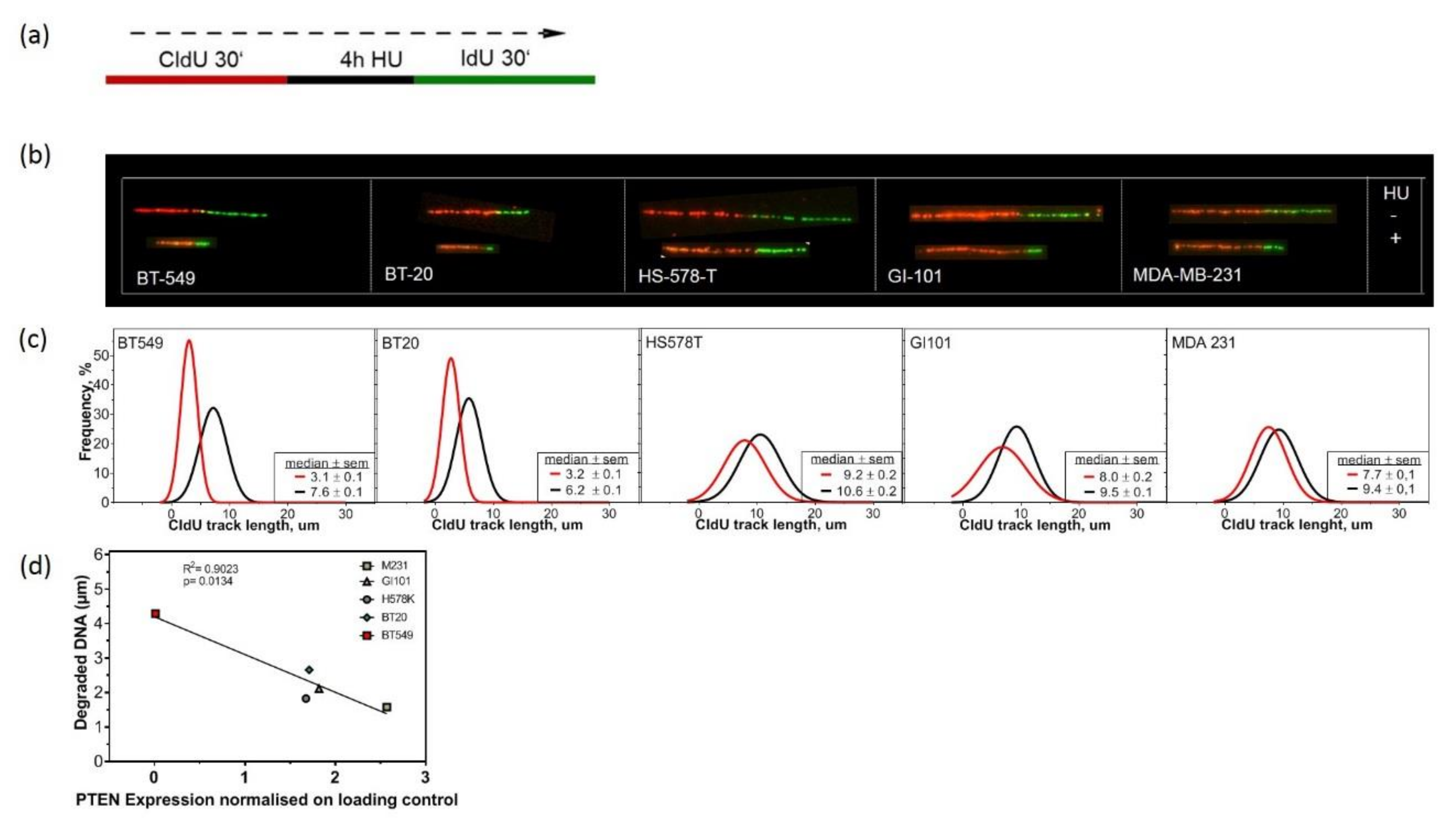
2.4. Lack of PTEN Leads to Replication Fork Instability in TNBC Cell Lines
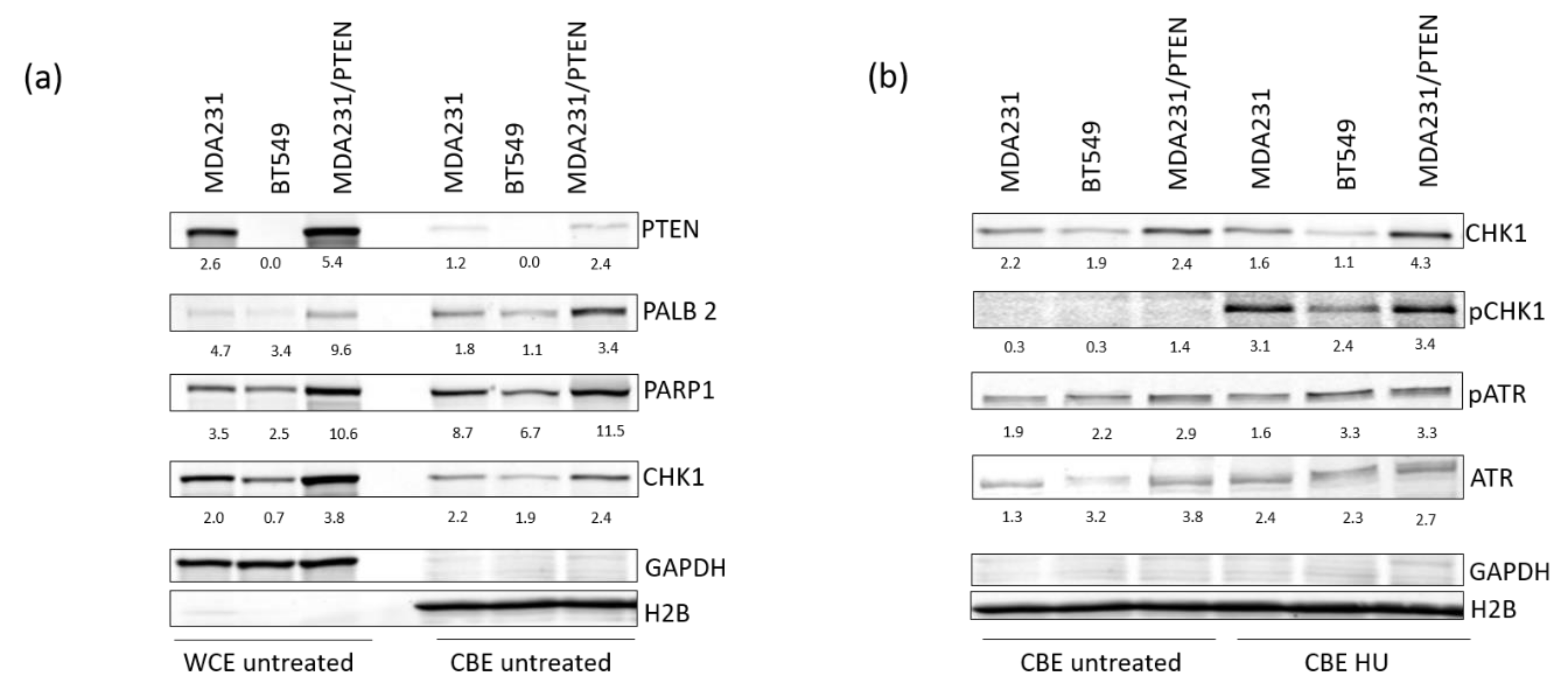
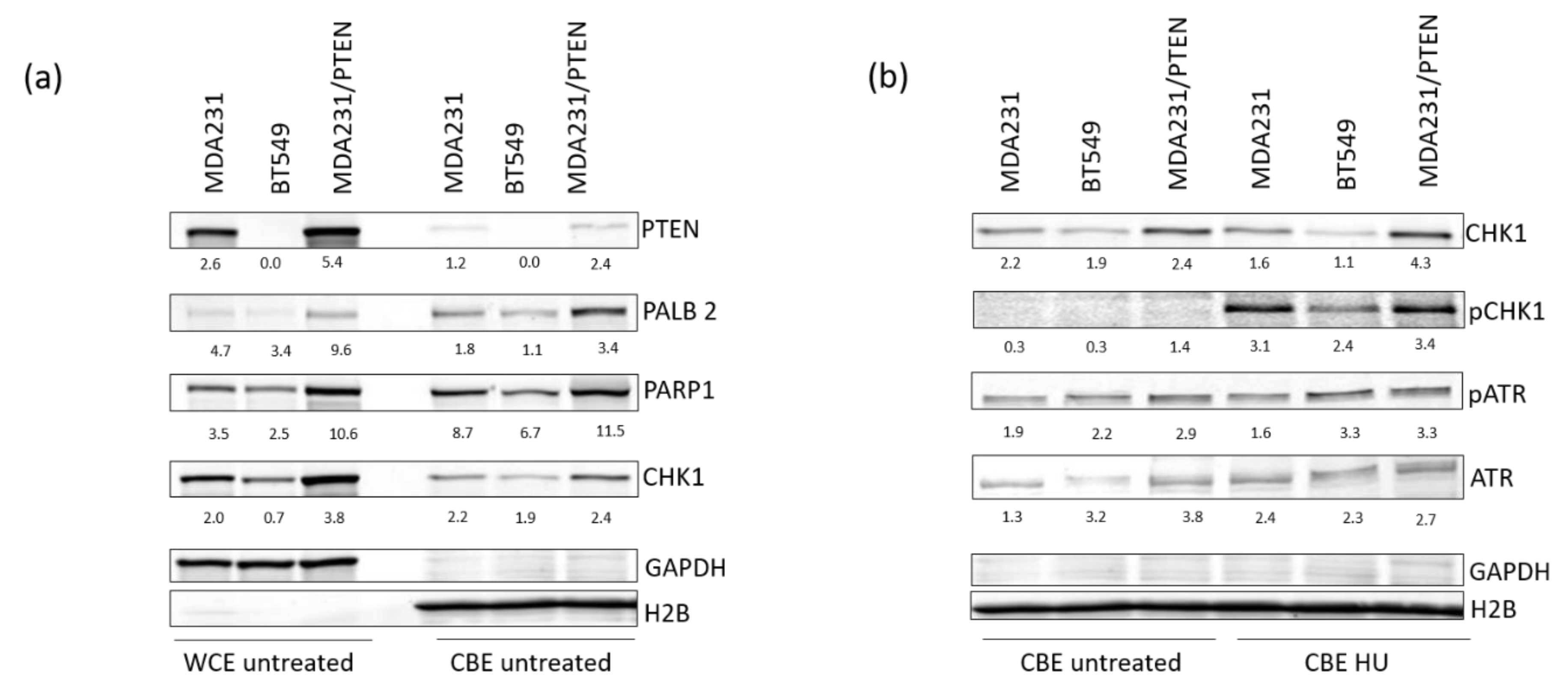
2.5. PTEN Protects Replication Forks by Supporting the Recruitment of Repair Proteins to the Chromatin
3. Discussion
4. Material and Methods
4.1. Cell Culture, Plasmids and Survival Assays
4.2. Clinical in Silico Analysis
4.3. Mutagenesis Assay
4.4. Western Blotting
4.5. DNA Fiber Assay
4.6. Data Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bayani, J.; Selvarajah, S.; Maire, G.; Vukovic, B.; Al-Romaih, K.; Zielenska, M.; Squire, J.A. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Semin. Cancer Biol. 2007, 17, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Funk, L.C.; Zasadil, L.M.; Weaver, B.A. Living in cin: Mitotic infidelity and its consequences for tumor promotion and suppression. Dev. Cell 2016, 39, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tijhuis, A.E.; Johnson, S.C.; McClelland, S.E. The emerging links between chromosomal instability (cin), metastasis, inflammation and tumour immunity. Mol. Cytogenet. 2019, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. Pten in the maintenance of genome integrity: From DNA replication to chromosome segregation. Bioessays 2017, 39, 1700082. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear pten in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [Green Version]
- McEllin, B.; Camacho, C.V.; Mukherjee, B.; Hahm, B.; Tomimatsu, N.; Bachoo, R.M.; Burma, S. Pten loss compromises homologous recombination repair in astrocytes: Implications for glioblastoma therapy with temozolomide or poly(adp-ribose) polymerase inhibitors. Cancer Res. 2010, 70, 5457–5464. [Google Scholar] [CrossRef] [Green Version]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear pten controls DNA repair and sensitivity to genotoxic stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of pten-assisted g2/m checkpoint impedes homologous recombination repair and enhances radio-curability and parp inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z.; Yang, J. Loss of pten expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef] [Green Version]
- Dean, S.J.; Perks, C.M.; Holly, J.M.; Bhoo-Pathy, N.; Looi, L.M.; Mohammed, N.A.; Mun, K.S.; Teo, S.H.; Koobotse, M.O.; Yip, C.H.; et al. Loss of pten expression is associated with igfbp2 expression, younger age, and late stage in triple-negative breast cancer. Am. J. Clin. Pathol. 2014, 141, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Siraj, A.K.; Prabhakaran, S.; Jehan, Z.; Ajarim, D.; Al-Dayel, F.; Tulbah, A.; Al-Kuraya, K.S. Loss of pten expression is associated with aggressive behavior and poor prognosis in middle eastern triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 151, 541–553. [Google Scholar] [CrossRef]
- Khan, F.; Esnakula, A.; Ricks-Santi, L.J.; Zafar, R.; Kanaan, Y.; Naab, T. Loss of pten in high grade advanced stage triple negative breast ductal cancers in african american women. Pathol. Res. Pract. 2018, 214, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Parp inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Exman, P.; Mallery, R.M.; Lin, N.U.; Parsons, H.A. Response to olaparib in a patient with germline brca2 mutation and breast cancer leptomeningeal carcinomatosis. NPJ Breast Cancer 2019, 5, 46. [Google Scholar] [CrossRef]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving therapeutic strategies to exploit chromosome instability in cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Parplys, A.C.; Seelbach, J.I.; Becker, S.; Behr, M.; Wrona, A.; Jend, C.; Mansour, W.Y.; Joosse, S.A.; Stuerzbecher, H.W.; Pospiech, H.; et al. High levels of rad51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired chk1 activation. Cell Cycle 2015, 14, 3190–3202. [Google Scholar] [CrossRef]
- Liu, C.; Srihari, S.; Lal, S.; Gautier, B.; Simpson, P.T.; Khanna, K.K.; Ragan, M.A.; Le Cao, K.A. Personalised pathway analysis reveals association between DNA repair pathway dysregulation and chromosomal instability in sporadic breast cancer. Mol. Oncol. 2016, 10, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Olziersky, A.M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 2019, 10, 3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to rad51-brca1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; Nishimura, R. A bone-seeking clone exhibits different biological properties from the mda-mb-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef]
- Hohensee, I.; Chuang, H.N.; Grottke, A.; Werner, S.; Schulte, A.; Horn, S.; Lamszus, K.; Bartkowiak, K.; Witzel, I.; Westphal, M.; et al. Pten mediates the cross talk between breast and glial cells in brain metastases leading to rapid disease progression. Oncotarget 2017, 8, 6155–6168. [Google Scholar] [CrossRef] [Green Version]
- Daboussi, F.; Courbet, S.; Benhamou, S.; Kannouche, P.; Zdzienicka, M.Z.; Debatisse, M.; Lopez, B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Parplys, A.C.; Zhao, W.; Sharma, N.; Groesser, T.; Liang, F.; Maranon, D.G.; Leung, S.G.; Grundt, K.; Dray, E.; Idate, R.; et al. Nucks1 is a novel rad51ap1 paralog important for homologous recombination and genome stability. Nucleic Acids Res. 2015, 43, 9817–9834. [Google Scholar] [CrossRef] [Green Version]
- Parplys, A.C.; Kratz, K.; Speed, M.C.; Leung, S.G.; Schild, D.; Wiese, C. Rad51ap1-deficiency in vertebrate cells impairs DNA replication. DNA Repair 2014, 24, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Nikkila, J.; Parplys, A.C.; Pylkas, K.; Bose, M.; Huo, Y.; Borgmann, K.; Rapakko, K.; Nieminen, P.; Xia, B.; Pospiech, H.; et al. Heterozygous mutations in palb2 cause DNA replication and damage response defects. Nat. Commun. 2013, 4, 2578. [Google Scholar] [CrossRef] [Green Version]
- Rehman, F.L.; Lord, C.J.; Ashworth, A. Synthetic lethal approaches to breast cancer therapy. Nat. Rev. Clin. Oncol. 2010, 7, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, C.; Lundin, C.; Helleday, T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001, 307, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Wurster, S.; Hennes, F.; Parplys, A.C.; Seelbach, J.I.; Mansour, W.Y.; Zielinski, A.; Petersen, C.; Clauditz, T.S.; Munscher, A.; Friedl, A.A.; et al. Parp1 inhibition radiosensitizes hnscc cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016, 7, 9732–9741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of pten mutant cells with parp inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Comen, E.A.; Robson, M. Poly(adp-ribose) polymerase inhibitors in triple-negative breast cancer. Cancer J. 2010, 16, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. Atr prohibits replication catastrophe by preventing global exhaustion of rpa. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, E.M.; Badie, S.; De Gregoriis, G.; Reislander, T.; Lai, X.; Porru, M.; Folio, C.; Moore, J.; Kopp, A.; Baguna Torres, J.; et al. Chlorambucil targets brca1/2-deficient tumours and counteracts parp inhibitor resistance. EMBO Mol. Med. 2019, 11, e9982. [Google Scholar] [CrossRef]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Conti, C.; Seiler, J.A.; Pommier, Y. The mammalian DNA replication elongation checkpoint: Implication of chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle 2007, 6, 2760–2767. [Google Scholar] [CrossRef]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, Y.; Wang, P.; Liang, H.; Cui, M.; Zhu, M.; Guo, L.; Su, Q.; Sun, Y.; McNutt, M.A.; et al. Pten regulates rpa1 and protects DNA replication forks. Cell Res. 2015, 25, 1189–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of rpa-ssdna complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for brca2 in blocking stalled replication fork degradation by mre11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by brca2 and parp1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. Parp1 and parp2 stabilise replication forks at base excision repair intermediates through fbh1-dependent rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Kruger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly(adp-ribose) binding to chk1 at stalled replication forks is required for s-phase checkpoint activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting atr in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rieckhoff, J.; Meyer, F.; Classen, S.; Zielinski, A.; Riepen, B.; Wikman, H.; Petersen, C.; Rothkamm, K.; Borgmann, K.; Parplys, A.C. Exploiting Chromosomal Instability of PTEN-Deficient Triple-Negative Breast Cancer Cell Lines for the Sensitization Against PARP1 Inhibition in a Replication-Dependent Manner. Cancers 2020, 12, 2809. https://doi.org/10.3390/cancers12102809
Rieckhoff J, Meyer F, Classen S, Zielinski A, Riepen B, Wikman H, Petersen C, Rothkamm K, Borgmann K, Parplys AC. Exploiting Chromosomal Instability of PTEN-Deficient Triple-Negative Breast Cancer Cell Lines for the Sensitization Against PARP1 Inhibition in a Replication-Dependent Manner. Cancers. 2020; 12(10):2809. https://doi.org/10.3390/cancers12102809
Chicago/Turabian StyleRieckhoff, Johanna, Felix Meyer, Sandra Classen, Alexandra Zielinski, Britta Riepen, Harriet Wikman, Cordula Petersen, Kai Rothkamm, Kerstin Borgmann, and Ann Christin Parplys. 2020. "Exploiting Chromosomal Instability of PTEN-Deficient Triple-Negative Breast Cancer Cell Lines for the Sensitization Against PARP1 Inhibition in a Replication-Dependent Manner" Cancers 12, no. 10: 2809. https://doi.org/10.3390/cancers12102809