The Promise of Circulating Tumor DNA (ctDNA) in the Management of Early-Stage Colon Cancer: A Critical Review


Simple Summary
Abstract
1. Introduction
2. The Biology of ctDNA
3. Methodological Considerations for the Use of ctDNA in Colon Cancer
4. Role of ctDNA in the Detection of Minimal Residual Disease (MRD)
5. Role of ctDNA in Assessing the Efficacy of Adjuvant Therapy
6. Potential Role of ctDNA in Surveillance
7. Limitations of ctDNA
8. Future Perspective and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Byrd, D.R.; Carducci, M.A.; Compton, C.C.; Fritz, A.G.; Greene, F.L. AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Ed.; AJCC: Chicago, IL, USA, 2017. [Google Scholar]
- Benson, A.I.; Venook, A.; Al-Hawary, M. NCCN Guidelines Version 1. 2020 Colon Cancer. Available online: https://wwwnccnorg/professionals/physician_gls/pdf/colonpdf (accessed on 8 August 2020).
- Bockelman, C.; Engelmann, B.E.; Kaprio, T.; Hansen, T.F.; Glimelius, B. Risk of recurrence in patients with colon cancer stage II and III: A systematic review and meta-analysis of recent literature. Acta Oncol. Stockh. Swed. 2015, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Påhlman, L.A.; Hohenberger, W.M.; Matzel, K.; Sugihara, K.; Quirke, P.; Glimelius, B. Should the Benefit of Adjuvant Chemotherapy in Colon Cancer Be Re-Evaluated? J. Clin. Oncol. 2016, 34, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346–392. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Sozzi, G.; Conte, D.; Mariani, L.; Lo Vullo, S.; Roz, L.; Lombardo, C.; Pierotti, M.A.; Tavecchio, L. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res. 2001, 61, 4675–4678. [Google Scholar] [PubMed]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Messaoudi, S.E.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Chen, C.; Hulbert, A.; Brock, M.V.; Yu, F. Non-blood circulating tumor DNA detection in cancer. Oncotarget 2017, 8, 69162–69173. [Google Scholar] [CrossRef] [PubMed]
- Emlen, W.; Mannik, M. Kinetics and mechanisms for removal of circulating single-stranded DNA in mice. J. Exp. Med. 1978, 147, 684–699. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, M.; Seong, M.W.; Kim, H.S.; Lee, Y.K.; Kang, H.J. Plasma vs. serum in circulating tumor DNA measurement: Characterization by DNA fragment sizing and digital droplet polymerase chain reaction. Clin. Chem. Lab Med. 2020, 58, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Alizadeh, A.; Adams, H.; Lee, J.; Klassen, S.; Palma, J. Early prediction of clinical outcomes in resected stage II and III colorectal cancer (CRC) through deep sequencing of circulating tumor DNA (ctDNA). J. Clin. Oncol. 2017, 35, 3591. [Google Scholar] [CrossRef]
- Sun, X.; Huang, T.; Cheng, F.; Huang, K.; Liu, M.; He, W.; Li, M.; Zhang, X.; Xu, M.; Chen, S.; et al. Monitoring colorectal cancer following surgery using plasma circulating tumor DNA. Oncol. Lett. 2018, 15, 4365–4375. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Scholer, L.V.; Reinert, T.; Orntoft, M.W.; Kassentoft, C.G.; Arnadottir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Chan, K.A.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer Genome Scanning in Plasma: Detection of Tumor-Associated Copy Number Aberrations, Single-Nucleotide Variants, and Tumoral Heterogeneity by Massively Parallel Sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef]
- Abbosh, C.; Swanton, C.; Birkbak, N.J. Clonal haematopoiesis: A source of biological noise in cell-free DNA analyses. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 358–359. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Peterson, C.Y.; Sriram, D.; Mahipal, A. Early stage colon cancer: Current treatment standards, evolving paradigms, and future directions. World J. Gastrointest Oncol. 2020, 12, 808–832. [Google Scholar] [CrossRef]
- Punt, C.J.A.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef]
- Nikbakht, H.; Jessa, S.; Sukhai, M.A.; Arseneault, M.; Zhang, T.; Letourneau, L.; Thomas, M.; Bourgey, M.; Roehrl, M.H.A.; Eveleigh, R.; et al. Latency and interval therapy affect the evolution in metastatic colorectal cancer. Sci. Rep. UK 2020, 10, 581. [Google Scholar] [CrossRef] [PubMed]
- Van Dessel, L.F.; Beije, N.; Helmijr, J.C.A.; Vitale, S.R.; Kraan, J.; Look, M.P.; de Wit, R.; Sleijfer, S.; Jansen, M.P.H.M.; Martens, J.W.M.; et al. Application of circulating tumor DNA in prospective clinical oncology trials—Standardization of preanalytical conditions. Mol. Oncol. 2017, 11, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Osterman, E.; Glimelius, B. Recurrence Risk After Up-to-Date Colon Cancer Staging, Surgery, and Pathology: Analysis of the Entire Swedish Population. Dis. Colon. Rectum. 2018, 61, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Boni, C.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C. Improved Overall Survival With Oxaliplatin, Fluorouracil, and Leucovorin As Adjuvant Treatment in Stage II or III Colon Cancer in the MOSAIC Trial. J. Clin. Oncol. 2009, 27, 3109–3116. [Google Scholar] [CrossRef]
- Parikh, A.; Van Seventer, E.; Boland, G. A plasma-only integrated genomic and epigenomic circulating tumor DNA (ctDNA) assay to inform recurrence risk in colorectal cancer (CRC). J. Clin. Oncol. 2019, 37, 3602. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Taieb, J.; Taly, V.; Vernerey, D.; Bourreau, C. LBA30_PR-Analysis of circulating tumor DNA (ctDNA) from patients enrolled in the IDEA-FRANCE phase III trial: Prognostic and predictive value for adjuvant treatment duration. Ann. Oncol. 2019, 30 (Suppl. 5), v851–v934. [Google Scholar] [CrossRef]
- Allegretti, M.; Cottone, G.; Carboni, F.; Cotroneo, E.; Casini, B.; Giordani, E.; Amoreo, C.A.; Buglioni, S.; Diodoro, M.; Pescarmona, E.; et al. Cross-sectional analysis of circulating tumor DNA in primary colorectal cancer at surgery and during post-surgery follow-up by liquid biopsy. J. Exp. Amp Clin. Cancer Res. CR 2020, 39, 69. [Google Scholar] [CrossRef]
- Maeda, H.; Kashiwabara, K.; Aoyama, T.; Oba, K.; Honda, M.; Mayanagi, S.; Kanda, M.; Hamada, C.; Sadahiro, S.; Sakamoto, J.; et al. Hazard rate of tumor recurrence over time in patients with colon cancer: Implications for postoperative surveillance from three Japanese Foundation for Multidisciplinary Treatment of Cancer (JFMC) clinical trials. J. Cancer 2017, 8, 4057–4064. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Welz, H.; Marienfeld, R.; Schmidt, S.A.; Kleger, A.; Ettrich, T.J.; Seufferlein, T. Treatment monitoring in metastatic colorectal cancer patients by quantification and KRAS genotyping of circulating cell-free DNA. PLoS ONE 2017, 12, e0174308. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Early change in circulating tumor DNA as a potential predictor of response to chemotherapy in patients with metastatic colorectal cancer. Sci. Rep. UK 2019, 9, 17358. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.; Wang, Y.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; Cho, J.H.; Faragher, I.; McKendrick, J.J.; et al. Serial circulating tumor DNA (ctDNA) analysis as a prognostic marker and a real-time indicator of adjuvant chemotherapy (CT) efficacy in stage III colon cancer (CC). J. Clin. Oncol. 2018, 36, 3516. [Google Scholar] [CrossRef]
- Elferink, M.A.G.; de Jong, K.P.; Klaase, J.M.; Siemerink, E.J.; de Wilt, J.H.W. Metachronous metastases from colorectal cancer: A population-based study in North-East Netherlands. Int. J. Colorectal Dis. 2015, 30, 205–212. [Google Scholar] [CrossRef]
- Snyder, R.A.; Hu, C.-Y.; Cuddy, A.; Francescatti, A.B.; Schumacher, J.R.; Van Loon, K.; You, Y.N.; Kozower, B.D.; Greenberg, C.C.; Schrag, D.; et al. Association Between Intensity of Posttreatment Surveillance Testing and Detection of Recurrence in Patients With Colorectal Cancer. JAMA 2018, 319, 2104–2115. [Google Scholar] [CrossRef]
- Jeffery, M.; Hickey, B.E.; Hider, P.N. Follow-up strategies for patients treated for non-metastatic colorectal cancer. Cochrane Database Syst. Rev. 2019, 9, CD002200. [Google Scholar] [CrossRef]
- Nicholson, B.D.; Shinkins, B.; Pathiraja, I.; Roberts, N.W.; James, T.J.; Mallett, S.; Perera, R.; Primrose, J.N.; Mant, D. Blood CEA levels for detecting recurrent colorectal cancer. Cochrane Database Syst. Rev. 2015, CD011134. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early stage NSCLC—Challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Springer, S.; Kinde, I.; Wong, H.-L.; Kosmider, S.; Tran, B.; Christie, M.; Thomson, B.N.; Wong, R.; et al. Serial circulating tumor DNA (ctDNA) and recurrence risk in patients (pts) with resectable colorectal liver metastasis (CLM). J. Clin. Oncol. 2016, 34, e15131. [Google Scholar] [CrossRef]
- Xie, H.; Mahoney, D.W.; Foote, P.H.; Burger, K.; Doering, K.; Taylor, W. Novel methylated DNA markers in plasma detect distant recurrence of colorectal cancer. J. Clin. Oncol. 2020, 38, 4088. [Google Scholar] [CrossRef]
- Libby, P.; Sidlow, R.; Lin, A.E.; Gupta, D.; Jones, L.W.; Moslehi, J.; Zeiher, A.; Jaiswal, S.; Schulz, C.; Blankstein, R.; et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Steensma, D.P. New Insights from Studies of Clonal Hematopoiesis. Clin. Cancer. Res. 2018, 24, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Severson, E.A.; Riedlinger, G.M.; Connelly, C.F.; Vergilio, J.A.; Goldfinger, M.; Ramkissoon, S.; Frampton, G.M.; Ross, J.S.; Fratella-Calabrese, A.; Gay, L.; et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood 2018, 131, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef]
- Grothey, A. Oxaliplatin-safety profile: Neurotoxicity. Semin. Oncol. 2003, 30, 5–13. [Google Scholar] [CrossRef]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N. Engl. J. Med. 2018, 378, 1177–1188. [Google Scholar] [CrossRef]
- Keppens, C.; Dequeker, E.M.C.; Patton, S.J.; Normanno, N.; Fenizia, F.; Butler, R.; Cheetham, M.; Fairley, J.A.; Williams, H.; Hall, J.A.; et al. International pilot external quality assessment scheme for analysis and reporting of circulating tumour DNA. BMC Cancer 2018, 18, 804. [Google Scholar] [CrossRef]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B.; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal–Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 1–14. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Gollerkeri, A.; Maharry, K.; et al. LBA-006BEACON CRC: A randomized, 3-Arm, phase 3 study of encorafenib and cetuximab with or without binimetinib vs. choice of either irinotecan or FOLFIRI plus cetuximab in BRAF V600E–mutant metastatic colorectal cancer. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Seymour, M.T.; Morton, D. FOxTROT: An international randomised controlled trial in 1052 patients (pts) evaluating neoadjuvant chemotherapy (NAC) for colon cancer. J. Clin. Oncol. 2019, 37, 3504. [Google Scholar] [CrossRef]
- Bhangu, J.; Beer, A.; Mittlboeck, M.; Tamandl, D.; Pulverer, W.; Taghizadeh, H.; Stremitzer, S.; Kaczirek, K.; Gruenberger, T.; Gnant, M.; et al. Circulating Free Methylated Tumor DNA Markers for Sensitive Assessment of Tumor Burden and Early Response Monitoring in Patients Receiving Systemic Chemotherapy for Colorectal Cancer Liver Metastasis. Ann. Surg. 2018, 268, 1. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Patient Population | ctDNA Assay Utilized | Blood Sample Collection Time Points | Major Findings | Other Relevant Findings |
|---|---|---|---|---|---|
| Tie et al. 2016 [10] | Stage II CC, n = 230 | Tumor-informed Safe-SeqS * | 4–10 weeks postop and every 3 months for up to 2 years | Cohort not receiving ACT. If positive ctDNA postop: 3-year RFS 0% (vs. 90% if ctDNA negative) and HR for recurrence 18 (95% CI, 7.9 to 40; p < 0.001). Cohort receiving ACT. If positive ctDNA post-ACT: HR for recurrence 11 (95% CI, 1.8 to 68; p = 0.001). | Median time interval between ctDNA detection and radiologic recurrence: 5.5 months (p = 0.001). |
| Scholer et al. 2017 [31] | Resected CRC, n = 45 (stages I to III, n = 21) | Tumor-informed ddPCR-based assay | Pre-op, days 8, 30, and every 3 months until month 36 | Localized CRC cohort: If positive ctDNA postop: HR for recurrence 37.7 (95% CI, 4.2–335.5; p < 0.001). 3-year RFS 0% vs. 73%. | ctDNA detection at 3 months after surgery predicted recurrence with an average lead time of 9.4 months compared to CT scans. |
| Diehn et al. 2017 [27] | Stages II and III CC, n = 145 | Tumor-informed CAPP-Seq | Single postop sample | Positive postop ctDNA: 2-year RFS 17% vs. 88% and HR for recurrence 10.3 (95% CI 2.3-46.9; p < 0.00001). | Monitoring multiple genomic alterations in the plasma improved sensitivity. |
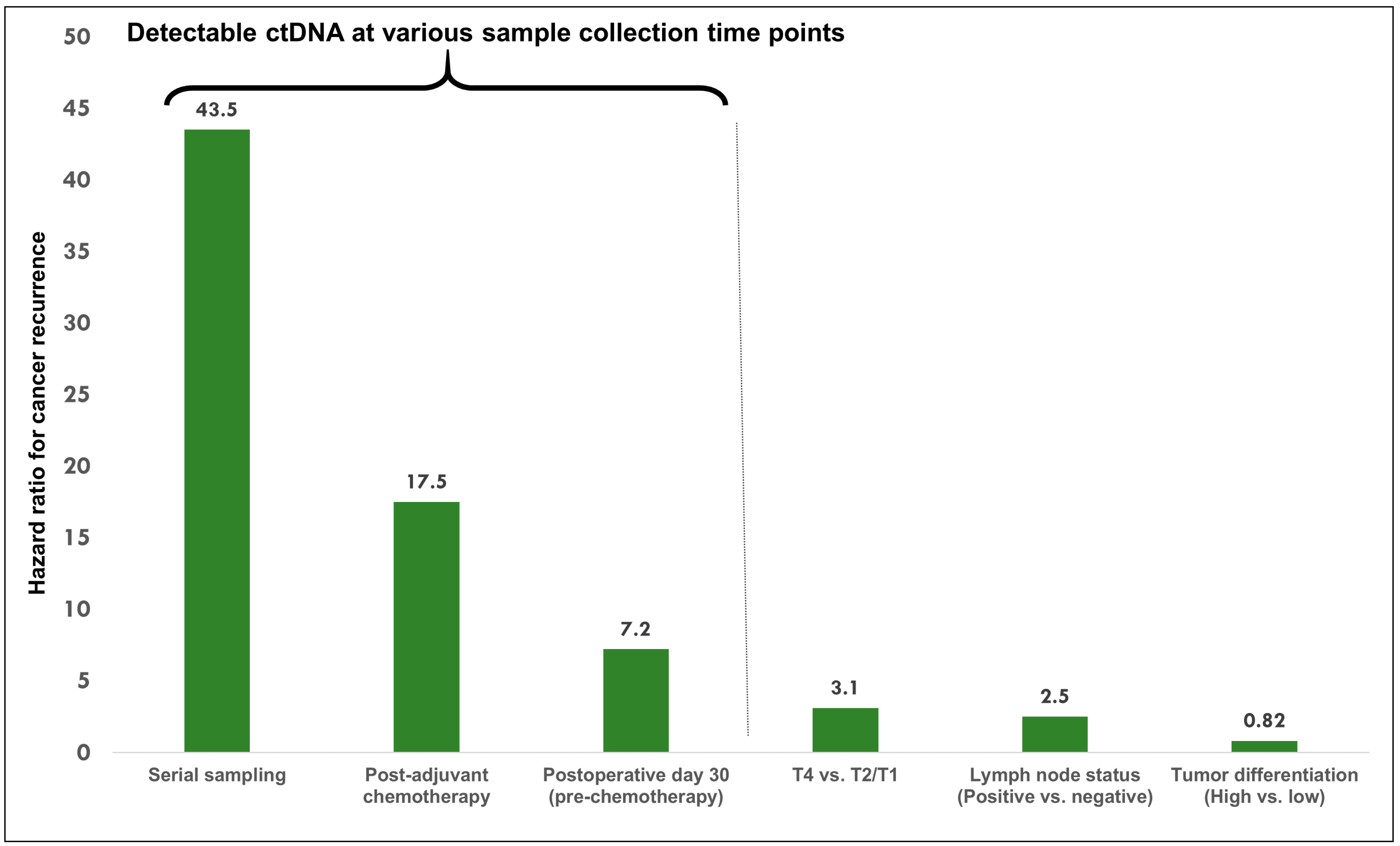
| Reinert et al. 2019 [7] | Stages I to III CRC, n = 130 | Tumor-informed, personalized, multiplex, PCR–based NGS ** | Preop, postop day 30, and every 3 months for up to 3 years. | HR for recurrence with positive ctDNA: Postop day 30: 7.2 (95% CI, 2.7-19.0; p < 0.001). Shortly after completion of ACT: 17.5 (95% CI, 5.4–56.5; p < 0.001). Serial monitoring post-ACT: 43.5 (95% CI, 9.8–193.5, p < 0.001). | Serial ctDNA analyses revealed disease recurrence up to 16.5 months ahead of radiologic imaging (mean, 8.7 months; range, 0.8–16.5 months). |
| Tie et al. 2019 [9] | Stage III CC, n = 96 | Tumor-informed Safe-SeqS * | 4–10 weeks postop and within 6 weeks of ACT completion | HR for recurrence with positive ctDNA: Postop: 7.5 on multivariable analysis (95% CI, 3.5–16.1; p < 0.001). Shortly after ACT: 6.8 (95% CI, 11.0-157.0; p < 0.001). | RFS at 3 years in patients with ctDNA positive vs. negative: Postop 47% vs. 76%, post-ACT 30% vs. 77%. |
| Tarazona et al. 2019 [8] | Stages I to III CC, n = 150 | Tumor-informed ddPCR | Preop, 6–8 weeks postop and every 4 months up to 5 years. | HR for recurrence with positive ctDNA: Postop (after multivariable adjustment):11.6 (95% CI, 3.6–36.8; p < 0.001). Post- ACT: 10.02 (95% CI, 9.2–307.3; p < 0.0001). | Positive ctDNA during surveillance preceded radiological recurrence with a median lead time of 11.5-months. |
| Parikh et al. 2019 [47] ¥ | Stages 0-IV CRC, n = 72. (Stage IV, n = 24) | Tumor-uninformed assay (LUNAR-1) | Postop and post-ACT | Postop positive ctDNA: HR for recurrence 8.7 (p < 0.0001), PPV 100%, NPV 76%. Post-ACT positive ctDNA: HR for recurrence 9.3 (p < 0.0001), PPV 100%, NPV 76% | Detection of ctDNA within 1 year of surgery predicted recurrence with a sensitivity of 69% and specificity of 94% |
| Wang et al. 2019 [48] | Stages I to III CC, n = 58 | Tumor-informed Safe-SeqS * | Postop at 1 month and then every 3–6 months | Recurrence rate among patients with positive ctDNA at any time point after surgery was 77% (10 of 13 patients). None of the 45 patients with negative ctDNA throughout the follow-up experienced a relapse (median follow-up 49 months). | Positive ctDNA preceded radiologic and clinical evidence of recurrence by a median of 3 months. |
| Study Identifier (Acronym) | Study Design | Study Population | Target Patient Number | ctDNA Assay Utilized | Study Description/Primary Endpoint |
|---|---|---|---|---|---|
| NCT04068103 (NRG GI005, COBRA) | Phase II/III | Resected stage IIA patients without high-risk features | 1408 | LUNAR-1 (Guardant Health) | Arm 1: Active surveillance. Arm 2: ctDNA directed therapy (ctDNA positive→mFOLFOX6/CAPOX for 6 months, ctDNA negative→active surveillance). The primary endpoints: Clearance of ctDNA with ACT (phase II) and RFS (phase III) |
| NCT04120701 (CIRCULATE) | Phase III | Resected Stage II patients | 1980 | Not reported | ctDNA positive→randomized (2:1) to receive ACT or no ACT. ctDNA negative→surveillance. Primary endpoint: 3-year DFS in ctDNA positive patients randomized to ACT or to follow-up. |
| ACTRN12615000381583 (DYNAMIC-II) | Phase III | Resected stage II patients | 450 | Safe-SeqS | Arm A: positive ctDNA→ACT, negative ctDNA→surveillance. Arm B: Treated at the discretion of the clinicians. Primary outcome measures-RFS and to evaluate whether a ctDNA guided adjuvant therapy strategy affects the number of patients treated with ACT. |
| ACTRN12617001566325 (DYNAMIC-III) | Phase II/III | Resected stage III patients | 1000 | Safe-SeqS | Arm A: Standard of care. Arm B: ctDNA informed (ctDNA negative→therapy de-escalation; ctDNA positive→therapy escalation). Primary endpoint: 3-year RFS (to demonstrate that a therapy de-escalation/escalation strategy informed by ctDNA is non-inferior to standard of care treatment). |
| NCT04084249 (IMPROVE-IT2) | Phase III | Resected high-risk stage II and stage III patients | 254 | Not reported | Randomization between ctDNA-guided surveillance and standard surveillance. Primary endpoint- Fraction of patients with relapse receiving curative-intent treatment |
| NCT03803553 | Phase III | Resected stage III patients | 500 | LUNAR-1 (Guardant Health) | Patients are enrolled after standard adjuvant chemotherapy in one of the 2 arms: 1. ctDNA negative: Surveillance; 2. ctDNA positive: (a) MSS patients→6 months of FOLFIRI vs. surveillance, (b) MSI high→6 months of nivolumab, (c) BRAF mutant/MSS→6 months of BRAF directed therapy. Primary outcome measures: 5-year DFS and clearance rate of ctDNA. |
| NCT04050345(TRACC) | Prospective, observational | Stage I, II and III patients with CRC | 1000 | Not reported | Multicenter, prospective study involving the collection and analysis of tumor tissue, serial blood samples, and clinical data in patients with newly diagnosed stage I, II and III CRC. Primary outcome measures: 1. The incidence of detectable ctDNA in patients with stage II and III CRC pre-operatively, 2. The correlation between detectable ctDNA at the first postoperative visit and DFS. |
| NCT04264702 (BESPOKE) | Prospective Observational study | Resected stage II and III colon cancer | 1000 | SIGNATERA™ | Patients will undergo a ctDNA testing following surgery and may be recommended for adjuvant chemotherapy or observation by their treating clinician. Follow up period- up to 2 years with periodic blood sample collection for ctDNA assay. Control arm will consist of matched stage II or stage III patients who have a minimum of 2 years of clinical follow-up data. Primary outcome measures: 1. To examine the impact of ctDNA on adjuvant treatment decisions. 2. To determine the rate of tumor recurrence while asymptomatic using ctDNA. |
| NCT04259944 (PEGASUS) | Phase II | Resected MSS stage III and high-risk stage II (T4N0) patients | 140 | LUNAR-1 (Guardant Health) | ctDNA guided ACT. (i) ctDNA positive→CAPOX for 3 months; (ii) ctDNA negative→capecitabine for 6 months but will be retested after 1 cycle, and if found ctDNA positive, will be switched to CAPOX. Primary outcome measure: Number of post-surgery and post-adjuvant false-negative cases after a double ctDNA-negative detection. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarti, S.; Xie, H.; Urrutia, R.; Mahipal, A. The Promise of Circulating Tumor DNA (ctDNA) in the Management of Early-Stage Colon Cancer: A Critical Review. Cancers 2020, 12, 2808. https://doi.org/10.3390/cancers12102808
Chakrabarti S, Xie H, Urrutia R, Mahipal A. The Promise of Circulating Tumor DNA (ctDNA) in the Management of Early-Stage Colon Cancer: A Critical Review. Cancers. 2020; 12(10):2808. https://doi.org/10.3390/cancers12102808
Chicago/Turabian StyleChakrabarti, Sakti, Hao Xie, Raul Urrutia, and Amit Mahipal. 2020. "The Promise of Circulating Tumor DNA (ctDNA) in the Management of Early-Stage Colon Cancer: A Critical Review" Cancers 12, no. 10: 2808. https://doi.org/10.3390/cancers12102808
APA StyleChakrabarti, S., Xie, H., Urrutia, R., & Mahipal, A. (2020). The Promise of Circulating Tumor DNA (ctDNA) in the Management of Early-Stage Colon Cancer: A Critical Review. Cancers, 12(10), 2808. https://doi.org/10.3390/cancers12102808