Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity


Simple Summary
Abstract

1. Introduction
2. Histone Methylation and Demethylation: An Historical Perspective
2.1. Molecular Mechanisms
2.2. Intersection with DNA Methylation
2.3. Developmental Roles
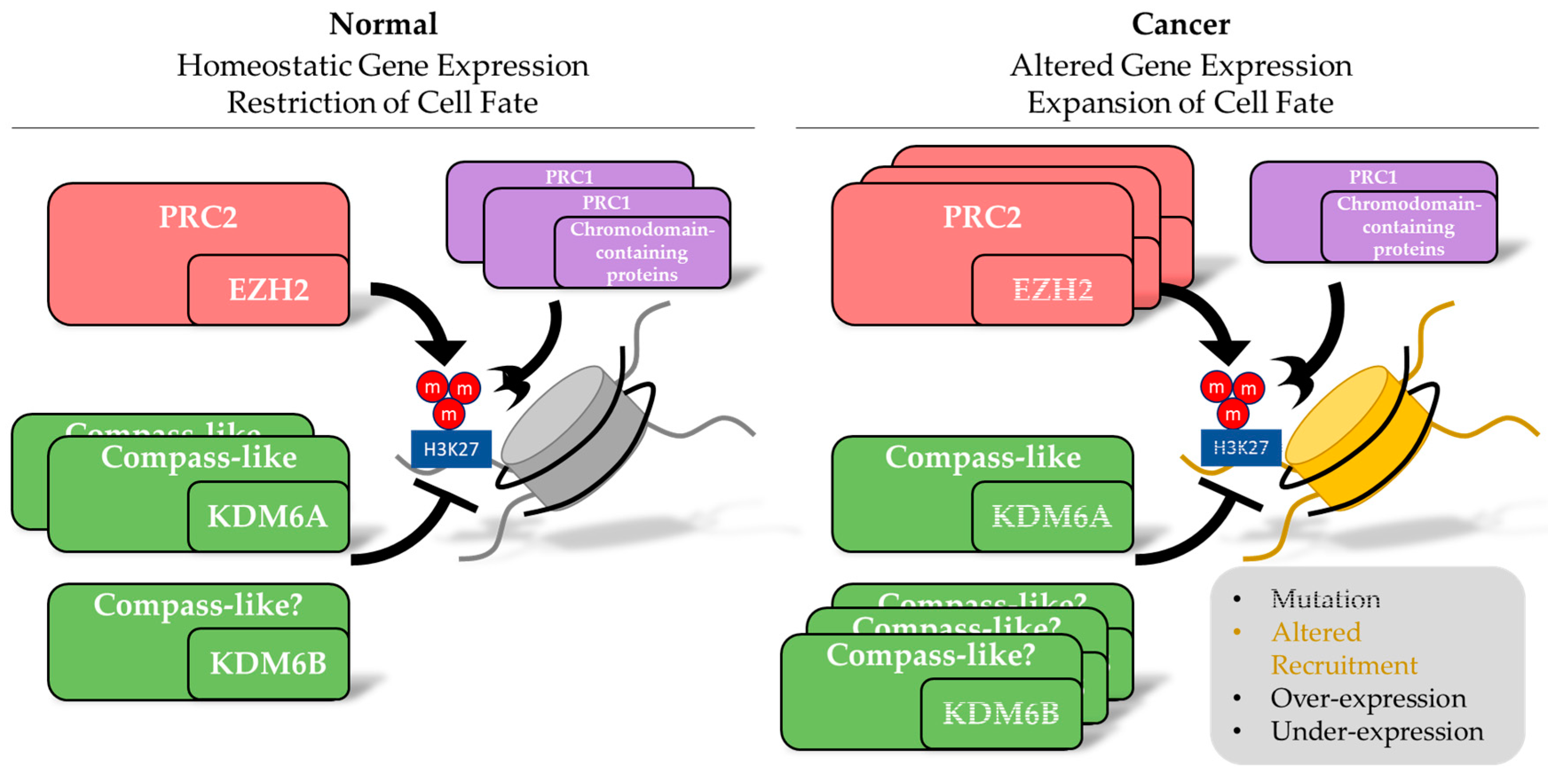
3. Alterations in Inducers of Methylation of H3K27 in Cancer

{kind=link}
{kind=link}
| EZH2 | ||||||
|---|---|---|---|---|---|---|
| Cancer Types with Evidence of Altered Expression or with Gene Altered in at Least 2% of Cases | Altered Expression | Cases in TCGA PAN-CANCER | ||||
| Amplification | Fusion | Deep Deletion | Mutation | % of Mutations Affecting the SET Domain | ||
| Prostate Adenocarcinoma | ↑ [64,66,81] | 0.80% | - | 0.20% | - | - |
| Invasive Breast Carcinoma | ↑ TCGA [82] | 0.65% | - | 0.46% | 0.28% | 100% |
| Colon Adenocarcinoma | ↑ [83,84] | - | - | - | 1.85% | 17% |
| Hepatocellular Carcinoma | ↑ [85] | 0.81% | - | 0.54% | 0.27% | 100% |
| Endometrial Carcinoma | ↑ [69] | 0.50% | - | - | 7.50% | 31% |
| Clear Cell Renal Carcinoma | ↑ [86] | 0.59% | - | - | 0.39% | 0% |
| Ovarian Epithelial | ↑ [TCGA] [87] | 7.00% | - | 0.30% | 0.30% | 0% |
| Melanoma | ↑ [69] | 2.00% | - | 0.20% | 4.70% | 21% |
| Sarcoma | ↑ [88,89] | 1.96% | - | 2.35% | 0.39% | 100% |
| Cervical Carcinoma | ↑ [90,91,92] | - | - | - | 4.35% | 0% |
| Lung Adenocarcinoma | ↑ [93] | 1.04% | 0.28% | 0.38% | 1.61% | 0% |
| Esophagogastric Adenocarcinoma | 0.19% | - | 1.56% | 1.17% | 0% | |
| Glioblastoma | ↑ TCGA ↓ [94] | 1.86% | - | - | 0.68% | 25% |
| Leukemia | ↑ [95] ↓CLL [96,97,98] | - | - | 1.00% | 1.00% | 25% |
| Bladder Urothelial Carcinoma | ↑ [99] | 0.49% | - | 0.49% | 1.46% | 40% |
| Adrenocortical Carcinoma | ↑ [100] | 1.10% | - | 1.10% | - | - |
| Esophageal Squamous Cell Carcinoma | ↑ [101] | 1.05% | - | 1.05% | - | - |
| Mature B-Cell Neoplasms | - | - | - | 2.08% | 0% | |
| Cholangiocarcinoma | - | - | - | 2.78% | 0% | |
4. KDM6 Family-Molecular Functions
5. Alterations in the Erasers of Methylation of H3K27 in Cancer
| KDM6A | ||||||
|---|---|---|---|---|---|---|
| Cancer Types with Evidence of Altered Expression or with Gene Altered in at Least 2% of Cases | Altered Expression | Cases in TCGA PAN-CANCER | ||||
| Amplification | Fusion | Deep Deletion | Mutation | % of Mutations Affecting the JmjC Domain | ||
| Bladder Urothelial Carcinoma | ↑ [184] | 0.49% | - | 2.68% | 25.55% | 74% |
| Esophageal Squamous Cell Carcinoma | ↓ [101] | 1.05% | - | 11.58% | 3.16% | 25% |
| Non-Small Cell Lung Cancer | 0.85% | 0.09% | 2.18% | 3.23% | 49% | |
| Endometrial Carcinoma | 0.85% | - | 0.68% | 6.66% | 22% | |
| Head and Neck Squamous Cell Carcinoma | ↓ [185] | 0.19% | - | 3.25% | 3.44% | 65% |
| Pancreatic Adenocarcinoma | - | - | 2.72% | 3.80% | 86% | |
| Stomach Adenocarcinoma | 0.68% | - | 1.82% | 3.41% | 40% | |
| Melanoma | ↓ [186] | - | - | 0.23% | 5.18% | 11% |
| Cervical Squamous Cell Carcinoma | 0.34% | - | 1.35% | 3.03% | 40% | |
| Prostate Adenocarcinoma | ↑ [187] | 0.20% | - | 1.21% | 2.02% | 25% |
| Mature B-Cell Neoplasms | 2.08% | - | - | 2.08% | 0% | |
| Ovarian Epithelial Tumor | ↓ [188] | 2.23% | 0.17% | 1.03% | 0.68% | 33% |
| Lung Adenocarcinoma | 0.88% | 0.18% | 0.71% | 1.94% | 31% | |
| Esophagogastric Adenocarcinoma | 0.97% | - | 1.95% | 3.50% | 0% | |
| Colorectal Adenocarcinoma | 0.17% | 0.51% | - | 2.53% | 33% | |
| Renal Non-Clear Cell Carcinoma | - | - | - | 3.16% | 0% | |
| Sarcoma | 1.18% | - | 0.78% | 0.39% | 25% | |
| Hepatocellular Carcinoma | 0.54% | - | 1.63% | 0.81% | 33% | |
| Invasive Breast Carcinoma | ↑ [189] | 0.65% | 0.09% | 0.18% | 1.94% | 0% |
| Leukemia | ↑ Adult T-cell [190] | 0.50% | - | 0.50% | 1.50% | 0% |
| Pleural Mesothelioma | - | - | 1.15% | 1.15% | 0% | |
| Cervical Adenocarcinoma | - | - | - | 2.17% | 29% | |
| KDM6B | ||||||
|---|---|---|---|---|---|---|
| Cancer Types with Evidence of Altered Expression or with Gene Altered in at Least 2% of Cases | Altered Expression | Cases in TCGA PAN-CANCER | ||||
| Amplification | Fusion | Deep Deletion | Mutation | % of Mutations Affecting the JmjC Domain | ||
| Papillary Renal Cell | ↑ [194] | - | - | 0.35% | 1.06% | 66% |
| Oligodendroglioma | ↑ [195] | - | - | - | - | - |
| Melanoma | - | - | 0.45% | 8.33% | 13% | |
| Endometrial carcinoma | 0.17% | - | 0.17% | 7.68% | 19% | |
| Miscellaneous Neuroepithelial Tumor | - | - | 3.23% | 3.23% | 0% | |
| Colorectal Adenocarcinoma | - | - | 1.18% | 4.21% | 38% | |
| Esophagogastric Adenocarcinoma | 0.19% | - | 0.58% | 4.09% | 0% | |
| Prostate Adenocarcinoma | ↑ [196] | - | - | 4.05% | 0.81% | 25% |
| Esophageal Squamous Cell Carcinoma | 1.05% | - | 1.05% | 2.11% | 50% | |
| Sarcoma | 1.57% | - | 1.18% | 0.78% | 0% | |
| Adrenocortical Carcinoma | - | - | 1.10% | 2.20% | 0% | |
| Hepatocellular Carcinoma | ↑ [197] | - | - | 2.44% | 0.54% | 0% |
| Non-Small Cell Lung Cancer | 0.09% | - | 0.76% | 1.99% | 0% | |
| Bladder Urothelial Carcinoma | - | - | 0.97% | 1.70% | 14% | |
| Cervical Squamous Cell Carcinoma | - | - | 0.40% | 1.99% | 0% | |
| Cervical Adenocarcinoma | - | - | 2.17% | - | - | |
| T-Cell Lymphomas | ↓ [198] | - | - | - | - | - |
| Mature B-Cell Neoplasms | ↓ [97] | - | - | 2.08% | - | - |
6. Alterations in the Substrate and Readers of Methylation of H3K27 and Alterations in DNA Methylation in Cancer
7. Cancer Cell Plasticity, EMT, and H3K27me3
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2020, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef]
- Baylin, B.S.; Jones, P.A. A decade of exploring the cancer epigenome–biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Feinberg, P.A.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- You, S.J.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, A.W.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, 6348. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.R.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar] [CrossRef]
- Raaphorst, F.M.; van Kemenade, F.J.; Blokzijl, T.; Fieret, E.; Hamer, K.M.; Satijn, D.P.; Otte, A.P.; Meijer, C.J. Coexpression of BMI-1 and EZH2 polycomb group genes in Reed-Sternberg cells of Hodgkin’s disease. Am. J. Pathol. 2000, 157, 709–715. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Völkel, P.; Dupret, B.; Le Bourhis, X.; Angrand, P.O. Diverse involvement of EZH2 in cancer epigenetics. Am. J. Transl. Res. 2015, 7, 175–193. [Google Scholar]
- Nekrasov, M.; Klymenko, T.; Fraterman, S.; Papp, B.; Oktaba, K.; Köcher, T.; Cohen, A.; Stunnenberg, H.G.; Wilm, M.; Müller, J. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. Embo. J. 2007, 26, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Lo, P.W.; Yeh, Y.H.; Hsu, P.H.; Peng, S.H.; Teng, Y.C.; Kang, M.L.; Wong, C.H.; Juan, L.J. O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. USA 2014, 111, 1355–1360. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef]
- Klose, J.R.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Canovas, S.; Cibelli, J.B.; Ross, P.J. Jumonji domain-containing protein 3 regulates histone 3 lysine 27 methylation during bovine preimplantation development. Proc. Natl. Acad. Sci. USA 2012, 109, 2400–2405. [Google Scholar] [CrossRef]
- Paik, K.W.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152. [Google Scholar] [CrossRef]
- Arifuzzaman, S.; Khatun, M.R.; Khatun, R. Emerging of lysine demethylases (KDMs): From pathophysiological insights to novel therapeutic opportunities. Biomed. Pharmacother. 2020, 129, 110392. [Google Scholar] [CrossRef]
- Klose, J.R.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Kooistra, M.S.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Tumber, A.; Che, K.; Cain, P.; Nowak, R.; Gileadi, C.; Oppermann, U. The roles of Jumonji-type oxygenases in human disease. Epigenomics 2014, 6, 89–120. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, T.M.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef]
- Statham, A.L.; Robinson, M.D.; Song, J.Z.; Coolen, M.W.; Stirzaker, C.; Clark, S.J. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res. 2012, 22, 1120–1127. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Lee, W.; Kim, J.; Yun, J.M.; Ohn, T.; Gong, Q. MeCP2 regulates gene expression through recognition of H3K27me3. Nat. Commun. 2020, 11, 3140. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The Polycomb-Group GeneEzh2 Is Required for Early Mouse Development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef]
- Schumacher, A.; Faust, C.; Magnuson, T. Positional cloning of a global regulator of anterior-posterior patterning in mice. Nature 1996, 384, 648. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. Embo. J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef]
- Liu, J.; An, L.; Wang, J.; Liu, Z.; Dai, Y.; Liu, Y.; Yang, L.; Du, F. Dynamic patterns of H3K4me3, H3K27me3, and Nanog during rabbit embryo development. Am. J. Transl. Res. 2019, 11, 430–441. [Google Scholar]
- Liu, X.; Wang, C.; Liu, W.; Li, J.; Li, C.; Kou, X.; Chen, J.; Zhao, Y.; Gao, H.; Wang, H.; et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 2016, 537, 558–562. [Google Scholar] [CrossRef]
- Zhang, B.; Zheng, H.; Huang, B.; Li, W.; Xiang, Y.; Peng, X.; Ming, J.; Wu, X.; Zhang, Y.; Xu, Q.; et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 2016, 537, 553–557. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell. 2016, 63, 1066–1079. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Malouf, G.G.; Taube, J.H.; Lu, Y.; Roysarkar, T.; Panjarian, S.; Estecio, M.R.; Jelinek, J.; Yamazaki, J.; Raynal, N.J.; Long, H.; et al. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013, 14, R144. [Google Scholar] [CrossRef]
- Van der Meulen, J.; Speleman, F.; Van Vlierberghe, P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics 2014, 9, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Nichol, J.N.; Dupere-Richer, D.; Ezponda, T.; Licht, J.D.; Miller, W.H., Jr. H3K27 Methylation: A Focal Point of Epigenetic Deregulation in Cancer. Adv. Cancer Res. 2016, 131, 59–95. [Google Scholar] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef]
- Friedman, J.M.; Liang, G.; Liu, C.C.; Wolff, E.M.; Tsai, Y.C.; Ye, W.; Zhou, X.; Jones, P.A. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009, 69, 2623–2629. [Google Scholar] [CrossRef]
- Saramaki, O.R.; Tammela, T.L.; Martikainen, P.M.; Vessella, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef]
- Wainwright, N.E.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef]
- Crea, F.; Paolicchi, E.; Marquez, V.E.; Danesi, R. Polycomb genes and cancer: Time for clinical application? Crit. Rev. Oncol. Hematol. 2012, 83, 184–193. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Hinz, S.; Kempkensteffen, C.; Christoph, F.; Hoffmann, M.; Krause, H.; Schrader, M.; Schostak, M.; Miller, K.; Weikert, S. Expression of the polycomb group protein EZH2 and its relation to outcome in patients with urothelial carcinoma of the bladder. J. Cancer Res. Clin. Oncol. 2008, 134, 331–336. [Google Scholar] [CrossRef]
- Wang, H.; Albadine, R.; Magheli, A.; Guzzo, T.J.; Ball, M.W.; Hinz, S.; Schoenberg, M.P.; Netto, G.J.; Gonzalgo, M.L. Increased EZH2 protein expression is associated with invasive urothelial carcinoma of the bladder. Urol. Oncol. 2012, 30, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Choe, M. Expression of EZH2 in renal cell carcinoma as a novel prognostic marker. Pathol. Int. 2012, 62, 735–741. [Google Scholar] [CrossRef]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.; Nilsson, J.; Mir, S.E.; van der Stoop, P.M.; Hulleman, E.; Niers, J.M.; de Witt Hamer, P.C.; Marquez, V.E.; Cloos, J.; Krichevsky, A.M.; et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget 2010, 1, 710–720. [Google Scholar] [CrossRef] [PubMed]
- McHugh, J.B.; Fullen, D.R.; Ma, L.; Kleer, C.G.; Su, L.D. Expression of polycomb group protein EZH2 in nevi and melanoma. J. Cutan. Pathol. 2007, 34, 597–600. [Google Scholar] [CrossRef]
- Li, X.; Gonzalez, M.E.; Toy, K.; Filzen, T.; Merajver, S.D.; Kleer, C.G. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am. J. Pathol. 2009, 175, 1246–1254. [Google Scholar] [CrossRef]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Ki, D.H.; Jeung, H.C.; Park, C.H.; Kang, S.H.; Lee, G.Y.; Lee, W.S.; Kim, N.K.; Chung, H.C.; Rha, S.Y. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int. J. Cancer 2007, 121, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Downey, T.; Eu, K.W.; Koh, P.K.; Cheah, P.Y. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin. Exp. Metastasis 2010, 27, 83–90. [Google Scholar] [CrossRef]
- Roessler, S.; Jia, H.L.; Budhu, A.; Forgues, M.; Ye, Q.H.; Lee, J.S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.Y.; Qin, L.X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef]
- Beroukhim, R.; Brunet, J.P.; Di Napoli, A.; Mertz, K.D.; Seeley, A.; Pires, M.M.; Linhart, D.; Worrell, R.A.; Moch, H.; Rubin, M.A.; et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69, 4674–4681. [Google Scholar] [CrossRef]
- Yoshihara, K.; Tajima, A.; Komata, D.; Yamamoto, T.; Kodama, S.; Fujiwara, H.; Suzuki, M.; Onishi, Y.; Hatae, M.; Sueyoshi, K.; et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009, 100, 1421–1428. [Google Scholar] [CrossRef]
- Detwiller, K.Y.; Fernando, N.T.; Segal, N.H.; Ryeom, S.W.; D’Amore, P.A.; Yoon, S.S. Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res. 2005, 65, 5881–5889. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef]
- Zhai, Y.; Kuick, R.; Nan, B.; Ota, I.; Weiss, S.J.; Trimble, C.L.; Fearon, E.R.; Cho, K.R. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007, 67, 10163–10172. [Google Scholar] [CrossRef]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Subramaniyam, S.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; Schneider, A.; Arias-Pulido, H.; et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol. Cancer 2008, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Arias-Pulido, H.; Subramaniyam, S.; Schneider, A.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes Chromosomes Cancer 2008, 47, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Chang, C.W.; Wu, Y.C.; Chen, K.C.; Lin, C.J.; Liang, S.C.; Lin, C.H.; Whang-Peng, J.; Hsu, S.L.; Chen, C.H.; et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 2007, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Haslinger, C.; Schweifer, N.; Stilgenbauer, S.; Döhner, H.; Lichter, P.; Kraut, N.; Stratowa, C.; Abseher, R. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J. Clin. Oncol. 2004, 22, 3937–3949. [Google Scholar] [CrossRef]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Béné, M.C.; De Vos, J.; Hernández, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef]
- Sanchez-Carbayo, M.; Socci, N.D.; Lozano, J.; Saint, F.; Cordon-Cardo, C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 2006, 24, 778–789. [Google Scholar] [CrossRef]
- Giordano, T.J.; Thomas, D.G.; Kuick, R.; Lizyness, M.; Misek, D.E.; Smith, A.L.; Sanders, D.; Aljundi, R.T.; Gauger, P.G.; Thompson, N.W.; et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am. J. Pathol. 2003, 162, 521–531. [Google Scholar] [CrossRef]
- Hu, N.; Clifford, R.J.; Yang, H.H.; Wang, C.; Goldstein, A.M.; Ding, T.; Taylor, P.R.; Lee, M.P. Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics 2010, 11, 576. [Google Scholar] [CrossRef] [PubMed]
- Bugler, J.; Kinstrie, R.; Scott, M.T.; Vetrie, D. Epigenetic Reprogramming and Emerging Epigenetic Therapies in CML. Front. Cell Dev. Biol. 2019, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Lund, K.; Adams, P.D.; Copland, M. EZH2 in normal and malignant hematopoiesis. Front. Cell Dev. Biol. 2019, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, D.; Imaizumi, Y.; Hasegawa, H.; Osaka, A.; Tsukasaki, K.; Choi, Y.L.; Mano, H.; Marquez, V.E.; Hayashi, T.; Yanagihara, K.; et al. Overexpression of Enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T-cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica 2011, 96, 712–719. [Google Scholar] [CrossRef]
- Beguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Herrera-Merchan, A.; Arranz, L.; Ligos, J.M.; de Molina, A.; Dominguez, O.; Gonzalez, S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat. Commun. 2012, 3, 623. [Google Scholar] [CrossRef]
- Mousavi, K.; Zare, H.; Wang, A.H.; Sartorelli, V. Polycomb Protein Ezh1 Promotes RNA Polymerase II Elongation. Mol. Cell 2012, 45, 255–262. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Wu, L.; Zhang, Q.; Yang, R.; Yang, Y.; Zhang, Z.; He, Q.; Chang, C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: Relation to adverse epigenetic alteration and poor prognostic scoring. Ann. Hematol. 2011, 90, 643–653. [Google Scholar] [CrossRef]
- Grubach, L.; Juhl-Christensen, C.; Rethmeier, A.; Olesen, L.H.; Aggerholm, A.; Hokland, P.; Østergaard, M. Gene expression profiling of Polycomb, Hox and Meis genes in patients with acute myeloid leukaemia. Eur. J. Haematol. 2008, 81, 112–122. [Google Scholar] [CrossRef]
- Fujii, S.; Tokita, K.; Wada, N.; Ito, K.; Yamauchi, C.; Ito, Y.; Ochiai, A. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene 2011, 30, 4118–4128. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Thu, K.L.; Aviel-Ronen, S.; Vucic, E.A.; Gazdar, A.F.; Lam, S.; Tsao, M.S.; Lam, W.L. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS ONE 2013, 8, e71670. [Google Scholar] [CrossRef]
- Koh, C.M.; Iwata, T.; Zheng, Q.; Bethel, C.; Yegnasubramanian, S.; De Marzo, A.M. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2011, 2, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef] [PubMed]
- Garipov, A.; Li, H.; Bitler, B.G.; Thapa, R.J.; Balachandran, S.; Zhang, R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Mol. Cancer Res. 2013, 11, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, E.V.; Revenko, A.S.; Gemo, A.T.; Andrews, N.P.; Tepper, C.G.; Zou, J.X.; Cardiff, R.D.; Borowsky, A.D.; Chen, H.W. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010, 70, 9402–9412. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef]
- Esposito, F.; Tornincasa, M.; Pallante, P.; Federico, A.; Borbone, E.; Pierantoni, G.M.; Fusco, A. Down-regulation of the miR-25 and miR-30d contributes to the development of anaplastic thyroid carcinoma targeting the polycomb protein EZH2. J. Clin. Endocrinol. Metab. 2012, 97, E710–E718. [Google Scholar] [CrossRef]
- Salvatori, B.; Iosue, I.; Djodji Damas, N.; Mangiavacchi, A.; Chiaretti, S.; Messina, M.; Padula, F.; Guarini, A.; Bozzoni, I.; Fazi, F.; et al. Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer 2011, 2, 585–592. [Google Scholar] [CrossRef]
- Lu, J.; He, M.L.; Wang, L.; Chen, Y.; Liu, X.; Dong, Q.; Chen, Y.C.; Peng, Y.; Yao, K.T.; Kung, H.F.; et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011, 71, 225–233. [Google Scholar] [CrossRef]
- Huang, S.D.; Yuan, Y.; Zhuang, C.W.; Li, B.L.; Gong, D.J.; Wang, S.G.; Zeng, Z.Y.; Cheng, H.Z. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol Cancer 2012, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Alajez, N.M.; Shi, W.; Hui, A.B.; Bruce, J.; Lenarduzzi, M.; Ito, E.; Yue, S.; O’Sullivan, B.; Liu, F.F. Enhancer of Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell Death Dis. 2010, 1, e85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Liao, Y.J.; Cai, M.Y.; Liu, Y.H.; Liu, T.H.; Chen, S.P.; Bian, X.W.; Guan, X.Y.; Lin, M.C.; Zeng, Y.X.; et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut 2012, 61, 278–289. [Google Scholar] [CrossRef]
- Luo, C.; Merz, P.R.; Chen, Y.; Dickes, E.; Pscherer, A.; Schadendorf, D.; Eichmüller, S.B. MiR-101 inhibits melanoma cell invasion and proliferation by targeting MITF and EZH2. Cancer Lett. 2013, 341, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Heath, E.; Chen, W.; Cher, M.L.; Powell, I.; Heilbrun, L.; Li, Y.; Ali, S.; Sethi, S.; Hassan, O.; et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS ONE 2012, 7, e33729. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Wan, Y.; Sun, G.; Shi, L.; Bao, X.; Wang, Z. Let-7a inhibits proliferation and induces apoptosis by targeting EZH2 in nasopharyngeal carcinoma cells. Oncol. Rep. 2012, 28, 2101–2106. [Google Scholar] [CrossRef]
- Qiu, S.; Huang, D.; Yin, D.; Li, F.; Li, X.; Kung, H.F.; Peng, Y. Suppression of tumorigenicity by microRNA-138 through inhibition of EZH2-CDK4/6-pRb-E2F1 signal loop in glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1832, 1697–1707. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, Z.; Tan, Z.; He, R.; Zeng, X.; Xie, Y.; Li, S.; Tang, G.; Tang, H.; He, X. MicroRNA-124 inhibits proliferation and induces apoptosis by directly repressing EZH2 in gastric cancer. Mol. Cell Biochem. 2014, 392, 153–159. [Google Scholar] [CrossRef]
- Luo, C.; Tetteh, P.W.; Merz, P.R.; Dickes, E.; Abukiwan, A.; Hotz-Wagenblatt, A.; Holland-Cunz, S.; Sinnberg, T.; Schittek, B.; Schadendorf, D.; et al. miR-137 inhibits the invasion of melanoma cells through downregulation of multiple oncogenic target genes. J. Investig. Dermatol. 2013, 133, 768–775. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, L.; Huo, X.S.; Yuan, J.H.; Xu, D.; Yuan, S.X.; Zhu, N.; Zhou, W.P.; Yang, G.S.; Wang, Y.Z.; et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 2011, 54, 1679–1689. [Google Scholar] [CrossRef]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013, 333, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Ott, H.M.; Graves, A.P.; Pappalardi, M.B.; Huddleston, M.; Halsey, W.S.; Hughes, A.M.; Groy, A.; Dul, E.; Jiang, Y.; Bai, Y.; et al. A687V EZH2 is a driver of histone H3 lysine 27 (H3K27) hypertrimethylation. Mol. Cancer Ther. 2014, 13, 3062–3073. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef]
- Richart, L.; Margueron, R. Drugging histone methyltransferases in cancer. Curr. Opin. Chem. Biol. 2020, 56, 51–62. [Google Scholar] [CrossRef]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in Myeloid Malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih Ie, M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Lu, Y.; Chen, W.; Juran, B.D.; Coltescu, C.; et al. Variants at IRF5-TNPO3, 17q12–21 and MMEL1 are associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 655–657. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef]
- He, W.; Cai, Q.; Sun, F.; Zhong, G.; Wang, P.; Liu, H.; Luo, J.; Yu, H.; Huang, J.; Lin, T. linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim. Biophys. Acta 2013, 1832, 1528–1537. [Google Scholar] [CrossRef]
- Cardoso, C.; Mignon, C.; Hetet, G.; Grandchamps, B.; Fontes, M.; Colleaux, L. The human EZH2 gene: Genomic organisation and revised mapping in 7q35 within the critical region for malignant myeloid disorders. Eur. J. Hum. Genet. 2000, 8, 174–180. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Chagraoui, J.; Krosl, J.; Gendron, P.; Wilhelm, B.; Lemieux, S.; Boucher, G.; Chagnon, P.; Drouin, S.; Lambert, R.; et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012, 26, 651–656. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E.; et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef]
- Smith, E.R.; Lee, M.G.; Winter, B.; Droz, N.M.; Eissenberg, J.C.; Shiekhattar, R.; Shilatifard, A. Drosophila UTX is a histone H3 Lys27 demethylase that colocalizes with the elongating form of RNA polymerase II. Mol. Cell. Biol. 2008, 28, 1041–1046. [Google Scholar] [CrossRef]
- Blatch, L.G.; Lässle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. Bioessays 1999, 21, 932–939. [Google Scholar] [CrossRef]
- Horton, J.R.; Gale, M.; Yan, Q.; Cheng, X. The Molecular Basis of Histone Demethylation, in DNA and Histone Methylation as Cancer Targets; Kaneda, A., Tsukada, Y.-I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 151–219. [Google Scholar]
- Hübner, R.M.; Spector, D.L. Role of H3K27 demethylases Jmjd3 and UTX in transcriptional regulation. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Hopkinson, R.J.; Vollmar, M.; Madden, S.K.; Gileadi, C.; Oppermann, U.; Schofield, C.J.; Johansson, C. Human UTY(KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J. Biol. Chem. 2014, 289, 18302–18313. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet 2012, 8, e1002964. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef]
- Issaeva, I.; Zonis, Y.; Rozovskaia, T.; Orlovsky, K.; Croce, C.M.; Nakamura, T.; Mazo, A.; Eisenbach, L.; Canaani, E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell Biol. 2007, 27, 1889–1903. [Google Scholar] [CrossRef]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef]
- Mohan, M.; Herz, H.M.; Smith, E.R.; Zhang, Y.; Jackson, J.; Washburn, M.P.; Florens, L.; Eissenberg, J.C.; Shilatifard, A. The COMPASS family of H3K4 methylases in Drosophila. Mol. Cell. Biol. 2011, 31, 4310–4318. [Google Scholar] [CrossRef]
- Vandamme, J.; Lettier, G.; Sidoli, S.; Di Schiavi, E.; Nørregaard Jensen, O.; Salcini, A.E. The C. elegans H3K27 demethylase UTX-1 is essential for normal development, independent of its enzymatic activity. PLoS Genet. 2012, 8, e1002647. [Google Scholar] [CrossRef]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [PubMed]
- Arcipowski, M.K.; Martinez, C.A.; Ntziachristos, P. Histone demethylases in physiology and cancer: A tale of two enzymes, JMJD3 and UTX. Curr. Opin. Genet. Dev. 2016, 36, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Perrigue, M.P.; Najbauer, J.; Barciszewski, J. Histone demethylase JMJD3 at the intersection of cellular senescence and cancer. Biochim. Biophys. Acta 2016, 1865, 237–244. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, J.; Sanghvi, V.; Mavrakis, K.; Durinck, K.; Fang, F.; Matthijssens, F.; Rondou, P.; Rosen, M.; Pieters, T.; Vandenberghe, P.; et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 2015, 125, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.M.; Madden, L.D.; Chen, Z.; Bolduc, C.; Buff, E.; Gupta, R.; Davuluri, R.; Shilatifard, A.; Hariharan, I.K.; Bergmann, A. The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol. Cell. Biol. 2010, 30, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Bullinger, L.; Basu, E.; Schlis, K.; Silverman, L.B.; Döhner, K.; Armstrong, S.A. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia 2012, 26, 1881–1883. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Dancik, G.M.; Im, K.M.; Edwards, M.G.; Turan, S.; Brown, J.; Ruiz-Rodriguez, C.; Owens, C.; Costello, J.C.; Guo, G.; et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin. Cancer Res. 2014, 20, 4935–4948. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Dyrskjøt, L.; Kruhøffer, M.; Thykjaer, T.; Marcussen, N.; Jensen, J.L.; Møller, K.; Ørntoft, T.F. Gene expression in the urinary bladder: A common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004, 64, 4040–4048. [Google Scholar] [CrossRef] [PubMed]
- Cromer, A.; Carles, A.; Millon, R.; Ganguli, G.; Chalmel, F.; Lemaire, F.; Young, J.; Dembélé, D.; Thibault, C.; Muller, D.; et al. Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 2004, 23, 2484–2498. [Google Scholar] [CrossRef] [PubMed]
- Talantov, D.; Mazumder, A.; Yu, J.X.; Briggs, T.; Jiang, Y.; Backus, J.; Atkins, D.; Wang, Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 2005, 11, 7234–7242. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Bonome, T.; Levine, D.A.; Shih, J.; Randonovich, M.; Pise-Masison, C.A.; Bogomolniy, F.; Ozbun, L.; Brady, J.; Barrett, J.C.; Boyd, J.; et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008, 68, 5478–5486. [Google Scholar] [CrossRef]
- Patani, N.; Jiang, W.G.; Newbold, R.F.; Mokbel, K. Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer Res. 2011, 31, 4115–4125. [Google Scholar]
- Choi, Y.L.; Tsukasaki, K.; O’Neill, M.C.; Yamada, Y.; Onimaru, Y.; Matsumoto, K.; Ohashi, J.; Yamashita, Y.; Tsutsumi, S.; Kaneda, R.; et al. A genomic analysis of adult T-cell leukemia. Oncogene 2007, 26, 1245–1255. [Google Scholar] [CrossRef]
- Manna, S.; Kim, J.K.; Baugé, C.; Cam, M.; Zhao, Y.; Shetty, J.; Vacchio, M.S.; Castro, E.; Tran, B.; Tessarollo, L.; et al. Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nat. Commun. 2015, 6, 8152. [Google Scholar] [CrossRef]
- Benyoucef, A.; Palii, C.G.; Wang, C.; Porter, C.J.; Chu, A.; Dai, F.; Tremblay, V.; Rakopoulos, P.; Singh, K.; Huang, S.; et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016, 30, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.D.; Eble, J.N.; Wang, M.; MacLennan, G.T.; Delahunt, B.; Brunelli, M.; Martignoni, G.; Lopez-Beltran, A.; Bonsib, S.M.; Ulbright, T.M.; et al. Molecular genetic evidence for the independent origin of multifocal papillary tumors in patients with papillary renal cell carcinomas. Clin. Cancer Res. 2005, 11, 7226–7233. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hui, A.M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006, 9, 287–300. [Google Scholar] [CrossRef]
- Hou, G.; Le Blancq, S.M.; E, Y.; Zhu, H.; Lee, M.G. Structure of a frequently rearranged rRNA-encoding chromosome in Giardia lamblia. Nucleic Acids Res. 1995, 23, 3310–3317. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Rossi, M.; Basso, K.; Zupo, S.; Went, P.; Klein, U.; Zinzani, P.L.; Baccarani, M.; et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J. Clin. Investig. 2007, 117, 823–834. [Google Scholar] [CrossRef]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.S.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef]
- Schulz, W.A.; Lang, A.; Koch, J.; Greife, A. The histone demethylase UTX/KDM6A in cancer: Progress and puzzles. Int. J. Cancer 2019, 145, 614–620. [Google Scholar] [CrossRef]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.K.; Tsai, M.C.; Poulin, G.; Adler, A.S.; Chen, S.; Liu, H.; Shi, Y.; Chang, H.Y. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 2010, 24, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sharma, A.; Dhar, S.S.; Lee, S.H.; Gu, B.; Chan, C.H.; Lin, H.K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Yomtoubian, S.; Lee, S.B.; Verma, A.; Izzo, F.; Markowitz, G.; Choi, H.; Cerchietti, L.; Vahdat, L.; Brown, K.A.; Andreopoulou, E.; et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 2020, 30, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef]
- Ene, C.I.; Edwards, L.; Riddick, G.; Baysan, M.; Woolard, K.; Kotliarova, S.; Lai, C.; Belova, G.; Cam, M.; Walling, J.; et al. Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS ONE 2012, 7, e51407. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 2014, 9, e96545. [Google Scholar] [CrossRef]
- Park, W.Y.; Hong, B.J.; Lee, J.; Choi, C.; Kim, M.Y. H3K27 Demethylase JMJD3 Employs the NF-κB and BMP Signaling Pathways to Modulate the Tumor Microenvironment and Promote Melanoma Progression and Metastasis. Cancer Res. 2016, 76, 161–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, L.; Stupack, D.G.; Bai, N.; Xun, J.; Ren, G.; Han, J.; Li, L.; Luo, Y.; Xiang, R.; et al. JMJD3 promotes survival of diffuse large B-cell lymphoma subtypes via distinct mechanisms. Oncotarget 2016, 7, 29387–29399. [Google Scholar] [CrossRef]
- Zhang, P.P.; Wang, X.L.; Zhao, W.; Qi, B.; Yang, Q.; Wan, H.Y.; Shuang, Z.Y.; Liu, M.; Li, X.; Li, S.; et al. DNA methylation-mediated repression of miR-941 enhances lysine (K)-specific demethylase 6B expression in hepatoma cells. J. Biol. Chem. 2014, 289, 24724–24735. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Wang, L.; Huang, X.; Lu, B.; Zou, C.; Wei, L.; Chu, J.; Eggers, P.K.; Chen, S.; Raston, C.L.; et al. Multifunctional nanoparticles for co-delivery of paclitaxel and carboplatin against ovarian cancer by inactivating the JMJD3-HER2 axis. Nanoscale 2017, 9, 13142–13152. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Yao, Q.; Wei, D.; Liu, M.; Geng, F.; Wang, Q.; Wang, Y.-s. KDM6B promotes ovarian cancer cell migration and invasion by induced transforming growth factor-β1 expression. J. Cell. Biochem. 2019, 120, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, D.Y.; Kim, M.-Y.; Kang, C. Synergistic triad epistasis of epigenetic H3K27me modifier genes, EZH2, KDM6A, and KDM6B, in gastric cancer susceptibility. Gastric Cancer 2019, 22, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, M.; Sheng, M.; Zhang, P.; Chen, Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2018, 144, 1065–1077. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef]
- Jin, Q.; Martinez, C.A.; Arcipowski, K.M.; Zhu, Y.; Gutierrez-Diaz, B.T.; Wang, K.K.; Johnson, M.R.; Volk, A.G.; Wang, F.; Wu, J.; et al. USP7 Cooperates with NOTCH1 to Drive the Oncogenic Transcriptional Program in T-Cell Leukemia. Clin. Cancer Res. 2019, 25, 222–239. [Google Scholar] [CrossRef]
- Sui, A.; Xu, Y.; Yang, J.; Pan, B.; Wu, J.; Guo, T.; Shen, Y.; Guo, X. The histone H3 Lys 27 demethylase KDM6B promotes migration and invasion of glioma cells partly by regulating the expression of SNAI1. Neurochem. Int. 2019, 124, 123–129. [Google Scholar] [CrossRef]
- Yin, X.; Yang, S.; Zhang, M.; Yue, Y. The role and prospect of JMJD3 in stem cells and cancer. Biomed. Pharmacother. 2019, 118, 109384. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers (Basel) 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Donson, A.; Foreman, N.K.; Dorris, K. H3 K27M Mutation in Gangliogliomas can be Associated with Poor Prognosis. Brain Pathol. 2017, 27, 846–850. [Google Scholar] [CrossRef]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallée, V.P.; Hébert, J.; Sauvageau, G. H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Weinberg, N.D.; Allis, C.D.; Lu, C. Oncogenic Mechanisms of Histone H3 Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026443. [Google Scholar] [CrossRef]
- Witt, O.; Albig, W.; Doenecke, D. Testis-specific expression of a novel human H3 histone gene. Exp. Cell Res. 1996, 229, 301–306. [Google Scholar] [CrossRef]
- Wiedemann, S.M.; Mildner, S.N.; Bönisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell. Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.R.; Tsai, S.; Bonner, W.M. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 1982, 31, 367–374. [Google Scholar] [CrossRef]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed]
- Drané, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Szenker, E.; Almouzni, G. Developmental roles of histone H3 variants and their chaperones. Trends Genet. 2013, 29, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Jacobsen, B.M.; Reiner, A.H.; Sørensen, A.L.; Küntziger, T.; Collas, P. Chromatin environment of histone variant H3.3 revealed by quantitative imaging and genome-scale chromatin and DNA immunoprecipitation. Mol. Biol. Cell. 2010, 21, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.M.; Yuen, B.T.; Barrilleaux, B.L.; Riggs, J.W.; O’Geen, H.; Cotterman, R.F.; Knoepfler, P.S. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenet. Chromatin. 2013, 6, 7. [Google Scholar] [CrossRef]
- Lin, J.C.; Conti, M.; Ramalho-Santos, M. Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development 2013, 140, 3624–3634. [Google Scholar] [CrossRef]
- Falkenberg, J.K.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.J.; Barretina, J.; Liu, J.; Howley, P.M.; et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Gui, B.; Xie, G.; Zhang, D.; Shang, Y.; Liang, J. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 2011, 286, 42414–42425. [Google Scholar] [CrossRef]
- Federico, A.; Pallante, P.; Bianco, M.; Ferraro, A.; Esposito, F.; Monti, M.; Cozzolino, M.; Keller, S.; Fedele, M.; Leone, V.; et al. Chromobox protein homologue 7 protein, with decreased expression in human carcinomas, positively regulates E-cadherin expression by interacting with the histone deacetylase 2 protein. Cancer Res. 2009, 69, 7079–7087. [Google Scholar] [CrossRef] [PubMed]
- Forzati, F.; Federico, A.; Pallante, P.; Abbate, A.; Esposito, F.; Malapelle, U.; Sepe, R.; Palma, G.; Troncone, G.; Scarfò, M.; et al. CBX7 is a tumor suppressor in mice and humans. J. Clin. Investig. 2012, 122, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Sepe, R.; Federico, A.; Forzati, F.; Bianco, M.; Fusco, A. CBX7 modulates the expression of genes critical for cancer progression. PLoS ONE 2014, 9, e98295. [Google Scholar] [CrossRef] [PubMed]
- Klauke, K.; Radulović, V.; Broekhuis, M.; Weersing, E.; Zwart, E.; Olthof, S.; Ritsema, M.; Bruggeman, S.; Wu, X.; Helin, K.; et al. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat. Cell Biol. 2013, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef]
- Score, J.; Hidalgo-Curtis, C.; Jones, A.V.; Winkelmann, N.; Skinner, A.; Ward, D.; Zoi, K.; Ernst, T.; Stegelmann, F.; Döhner, K.; et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012, 119, 1208–1213. [Google Scholar] [CrossRef]
- Ueda, T.; Sanada, M.; Matsui, H.; Yamasaki, N.; Honda, Z.I.; Shih, L.Y.; Mori, H.; Inaba, T.; Ogawa, S.; Honda, H. EED mutants impair polycomb repressive complex 2 in myelodysplastic syndrome and related neoplasms. Leukemia 2012, 26, 2557–2560. [Google Scholar] [CrossRef]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef]
- Keats, J.J.; Reiman, T.; Maxwell, C.A.; Taylor, B.J.; Larratt, L.M.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [Google Scholar] [CrossRef]
- Ezponda, T.; Popovic, R.; Shah, M.Y.; Martinez-Garcia, E.; Zheng, Y.; Min, D.J.; Will, C.; Neri, A.; Kelleher, N.L.; Yu, J.; et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013, 32, 2882–2890. [Google Scholar] [CrossRef]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Oyer, J.A.; Huang, X.; Zheng, Y.; Shim, J.; Ezponda, T.; Carpenter, Z.; Allegretta, M.; Okot-Kotber, C.I.; Patel, J.P.; Melnick, A.; et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014, 28, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.E.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Wu, T.; Fu, H.; Dai, C.; Wu, H.; Liu, N.; Li, X.; Xu, M.; Zhang, Z.; Niu, T.; et al. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 2012, 337, 971–975. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- van der Vlag, J.; Otte, A.P. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 1999, 23, 474–478. [Google Scholar] [CrossRef]
- Song, C.; Pan, X.; Ge, Z.; Gowda, C.; Ding, Y.; Li, H.; Li, Z.; Yochum, G.; Muschen, M.; Li, Q.; et al. Epigenetic regulation of gene expression by Ikaros, HDAC1 and Casein Kinase II in leukemia. Leukemia 2016, 30, 1436–1440. [Google Scholar] [CrossRef]
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell 2015, 57, 662–673. [Google Scholar] [CrossRef]
- Sinha, S.; Thomas, D.; Yu, L.; Gentles, A.J.; Jung, N.; Corces-Zimmerman, M.R.; Chan, S.M.; Reinisch, A.; Feinberg, A.P.; Dill, D.L.; et al. Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood 2015, 125, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Behm, F.G.; Raimondi, S.C.; Frestedt, J.L.; Liu, Q.; Crist, W.M.; Downing, J.R.; Rivera, G.K.; Kersey, J.H.; Pui, C.H. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood 1996, 87, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, E.; Zuber, J.; Rappaport, A.; Taylor, M.; Johns, C.; Lowe, S.W.; Vakoc, C.R. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene 2013, 32, 930–938. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Zhang, M.T.; Huang, T.; Wang, R.F. Cross talk of chromosome instability, CpG island methylator phenotype and mismatch repair in colorectal cancer. Oncol. Lett. 2018, 16, 1736–1746. [Google Scholar] [CrossRef]
- Weisenberger, D.J. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 10–522. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; Thompson, C.B.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.G.; Barwick, B.G.; Fau-Jin, G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.-T.; Bigner, D.D.; Vertino, P.M.; et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Cohen, L.A.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell. Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell. 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Davis, L.R.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Vicente-Duenas, C.; Gutierrez de Diego, J.; Rodriguez, F.D.; Jimenez, R.; Cobaleda, C. The role of cellular plasticity in cancer development. Curr. Med. Chem. 2009, 16, 3676–3685. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Taube, J.H.; Malouf, G.G.; Lu, E.; Sphyris, N.; Vijay, V.; Ramachandran, P.P.; Ueno, K.R.; Gaur, S.; Nicoloso, M.S.; Rossi, S.; et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci. Rep. 2013, 3, 2687. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Jordan, N.V.; Prat, A.; Abell, A.N.; Zawistowski, J.S.; Sciaky, N.; Karginova, O.A.; Zhou, B.; Golitz, B.T.; Perou, C.M.; Johnson, G.L. SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c controls epithelial-mesenchymal transition by inducing Wnt5a signaling. Mol. Cell Biol. 2013, 33, 3011–3025. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Specht, S.; Lunz, J.G., 3rd; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. Cooperation of p300 and PCAF in the control of microRNA 200c/141 transcription and epithelial characteristics. PLoS ONE 2012, 7, e32449. [Google Scholar] [CrossRef]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Jin, F.Y.; Lu, K.; Wan, L.; Xie, M.; Xu, T.P.; De, W.; Wang, Z.X. SUZ12 promotes gastric cancer cell proliferation and metastasis by regulating KLF2 and E-cadherin. Tumour. Biol. 2015, 36, 5341–5351. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [PubMed]
- Collett, K.; Eide, G.E.; Arnes, J.; Stefansson, I.M.; Eide, J.; Braaten, A.; Aas, T.; Otte, A.P.; Akslen, L.A. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin. Cancer Res. 2006, 12, 1168–1174. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Puppe, J.; Drost, R.; Liu, X.; Joosse, S.A.; Evers, B.; Cornelissen-Steijger, P.; Nederlof, P.; Yu, Q.; Jonkers, J.; van Lohuizen, M.; et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009, 11, R63. [Google Scholar] [CrossRef]
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef]
- Martin, A.; Cano, A. Tumorigenesis: Twist1 links EMT to self-renewal. Nat. Cell Biol. 2010, 12, 924–925. [Google Scholar] [CrossRef]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Dhar, S.S.; Lee, S.H.; Chen, K.; Zhu, G.; Oh, W.; Allton, K.; Gafni, O.; Kim, Y.Z.; Tomoiga, A.S.; Barton, M.C.; et al. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 2016, 44, 3659–3674. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, S.; Chen, X.; Wang, C.Y. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef] [PubMed]
- Serresi, M.; Gargiulo, G.; Proost, N.; Siteur, B.; Cesaroni, M.; Koppens, M.; Xie, H.; Sutherland, K.D.; Hulsman, D.; Citterio, E.; et al. Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 2016, 29, 17–31. [Google Scholar] [CrossRef]
- Markopoulos, G.S.; Roupakia, E.; Marcu, K.B.; Kolettas, E. Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation. Cells 2019, 8, 1143. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef]
- Zha, L.; Cao, Q.; Cui, X.; Li, F.; Liang, H.; Xue, B.; Shi, H. Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med. Oncol. 2016, 33, 21. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, J.H.; Park, M.; Won, H.Y.; Joo, H.S.; Lee, C.H.; Lee, J.Y.; Kong, G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 2015, 16, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.N.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Xu, L.; Wu, X.; Zhang, L.; Fei, X.; Cao, Y.; Zhang, F. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp. Cell Res. 2017, 359, 405–414. [Google Scholar] [CrossRef]
- Terashima, M.; Ishimura, A.; Wanna-Udom, S.; Suzuki, T. Epigenetic regulation of epithelial-mesenchymal transition by KDM6A histone demethylase in lung cancer cells. Biochem Biophys.. Res. Commun. 2017, 490, 1407–1413. [Google Scholar] [CrossRef]
- van den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of Histone Demethylases LSD1 and UTX Regulates ERalpha Signaling in Breast Cancer. Cancers (Basel) 2019, 11, 2027. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular Plasticity in Breast Cancer Progression and Therapy. Front. Mol. Biosci. 2020, 7, 72. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Dekker, J.; Misteli, T. Long-Range Chromatin Interactions. Cold Spring Harb. Perspect. Biol. 2015, 7, a019356. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell. 2016, 62, 668–680. [Google Scholar] [CrossRef]
- Allis, D.C.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes; Routledge: London, UK, 2014. [Google Scholar]
- Brock, A.; Chang, H.; Huang, S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet. 2009, 10, 336–342. [Google Scholar] [CrossRef]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, P.; Taube, J.H. Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers 2020, 12, 2792. https://doi.org/10.3390/cancers12102792
Das P, Taube JH. Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers. 2020; 12(10):2792. https://doi.org/10.3390/cancers12102792
Chicago/Turabian StyleDas, Provas, and Joseph H. Taube. 2020. "Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity" Cancers 12, no. 10: 2792. https://doi.org/10.3390/cancers12102792
APA StyleDas, P., & Taube, J. H. (2020). Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers, 12(10), 2792. https://doi.org/10.3390/cancers12102792