Target Enrichment Enables the Discovery of lncRNAs with Somatic Mutations or Altered Expression in Paraffin-Embedded Colorectal Cancer Samples

 , ,
, ,
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Design of a lncRNA-Focused Enrichment
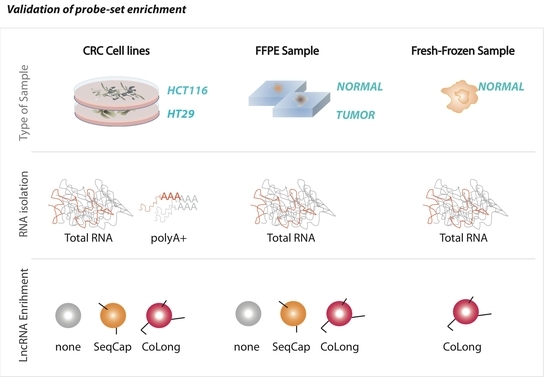
2.2. Validation of Probe-Based Enrichment of lncRNAs
2.3. Identification of Differentially Expressed lncRNAs in CRC
2.4. Validation of Gene Expression Data of a Subset of DE lncRNAs
2.5. Identification of CRC-Related Somatic Mutations in FFPE Samples
3. Material and Methods
3.1. Ethics Statement
3.2. Patients and Samples
3.3. RNA Isolation From FFPE, FF, and Cell Line Cultures
3.4. Custom Design of Probes for lncRNA Enrichment
3.5. Library Preparation, lncRNA Enrichment and Sequencing
3.6. Quality Control, Pseudo-Mapping and Gene Abundance Quantification
3.7. Differential Expression Analysis
3.8. Gene Expression Quantification of TCGA Samples
3.9. Characterization of Differentially-Expressed lncRNAs
3.10. SNP Calling
3.11. Validation of lncRNA Expression by Real-Time Quantitative PCR (RT-qPCR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, C.J.; Hendrich, B.; Rupert, J.L.; Lafrenière, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Clemson, C.M.; A McNeil, J.; Willard, H.F.; Lawrence, J.B. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Boil. 1996, 132, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martín, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.J.; Tazon-Vega, B.; Regev, A.; Rinn, J. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2016, 17, 135–143. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef]
- Du, Z.; Fei, T.; Verhaak, R.G.; Su, Z.; Zhang, Y.; Brown, M.; Chen, Y.; Liu, X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Boil. 2013, 20, 908–913. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Marguerat, S.; Bähler, J. RNA-seq: from technology to biology. Cell. Mol. Life Sci. 2009, 67, 569–579. [Google Scholar] [CrossRef]
- Farragher, S.M.; Tanney, A.; Kennedy, R.D.; Harkin, D.P. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem. Cell Boil. 2008, 130, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Abdueva, D.; Wing, M.; Schaub, B.; Triche, T.J.; Davicioni, E. Quantitative Expression Profiling in Formalin-Fixed Paraffin-Embedded Samples by Affymetrix Microarrays. J. Mol. Diagn. 2010, 12, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, B.; Sheng, Q.; Seitz, R.S.; Lawrence, K.; Morris, S.W.; Thomas, L.R.; Hout, D.R.; Schweitzer, B.L.; Guo, Y.; Pietenpol, J.A.; et al. Comparison of triple-negative breast cancer molecular subtyping using RNA from matched fresh-frozen versus formalin-fixed paraffin-embedded tissue. BMC Cancer 2017, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, J.; Thorsen, K.; Lund, M.K.; Hein, A.-M.K.; Hamilton-Dutoit, S.; Vang, S.; Nordentoft, I.; Birkenkamp-Demtröder, K.; Kruhøffer, M.; Hager, H.; et al. Next-Generation Sequencing of RNA and DNA Isolated from Paired Fresh-Frozen and Formalin-Fixed Paraffin-Embedded Samples of Human Cancer and Normal Tissue. PLoS ONE 2014, 9, e98187. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Sato, T.; Ono, T.; Okada, K.; Masuda, Y.; Ito, H. Malignant Mesotheliomas in Kure City, Japan: The Relationship of Asbestos Exposure. Cancer Investig. 1989, 7, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Conley, A.; Zhang, H.; Kim, H.L. Whole-Transcriptome profiling of formalin-fixed, paraffin-embedded renal cell carcinoma by RNA-seq. BMC Genom. 2014, 15, 1087. [Google Scholar] [CrossRef]
- Turnbull, A.K.; Selli, C.; Martínez-Pérez, C.; Fernando, A.; Renshaw, L.; Keys, J.; Figueroa, J.D.; He, X.; Tanioka, M.; Munro, A.F.; et al. Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: comparison of gene expression profiling approaches. BMC Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Kwong, L.N.; De Macedo, M.P.; Haydu, L.; Joon, A.Y.; Tetzlaff, M.T.; Calderone, T.L.; Wu, C.-J.; Kwong, M.K.; Roszik, J.; Hess, K.R.; et al. Biological Validation of RNA Sequencing Data From Formalin-Fixed Paraffin-Embedded Primary Melanomas. JCO Precis. Oncol. 2018, 2018, 1–19. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Clark, M.B.; Mercer, T.R.; Bussotti, G.; Leonardi, T.; Haynes, K.R.; Crawford, J.; E Brunck, M.; Cao, K.-A.L.; Thomas, G.P.; Chen, W.Y.; et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 2015, 12, 339–342. [Google Scholar] [CrossRef]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Carbonell, S.; Pérez-Lluch, S.; Abad, A.; Davis, C.; Gingeras, T.R.; Frankish, A.; Harrow, J.; Guigó, R.; et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat. Genet. 2017, 49, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Chugh, R.; Wu, Y.-M.; Wu, M.; Brennan, C.; Lonigro, R.; Su, F.; Wang, R.; Siddiqui, J.; Mehra, R.; et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015, 25, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Siebold, A.P.; Livi, C.B.; Lucas, A.B. SureSelectXT RNA Direct: A Technique for Expression Analysis Through Sequencing of Target-Enriched FFPE Total RNA. Breast Cancer 2018, 1783, 81–104. [Google Scholar] [CrossRef]
- Li, J.; Fu, C.; Speed, T.P.; Wang, W.; Symmans, W.F. Accurate RNA Sequencing From Formalin-Fixed Cancer Tissue to Represent High-Quality Transcriptome From Frozen Tissue. JCO Precis. Oncol. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Tan, J.C.; Bouriakov, V.D.; Feng, L.; A Richmond, T.; Burgess, D. Targeted LncRNA Sequencing with the SeqCap RNA Enrichment System. Advanced Structural Safety Studies 2016, 1402, 73–100. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Lynch, H.; De La Chapelle, A. Hereditary Colorectal Cancer. New Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Jass, J.R. Colorectal Cancer: A Multipathway Disease. Crit. Rev. Oncog. 2006, 12, 273–287. [Google Scholar] [CrossRef]
- Saus, E.; Brunet-Vega, A.; Iraola-Guzmán, S.; Pegueroles, C.; Gabaldón, T.; Pericay, C. Long Non-Coding RNAs As Potential Novel Prognostic Biomarkers in Colorectal Cancer. Front. Genet. 2016, 7, 54. [Google Scholar] [CrossRef]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Patel, D.; Rastan, S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991, 351, 329–331. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Martianov, I.; Ramadass, A.; Barros, A.S.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The Nuclear-Retained Noncoding RNA MALAT1 Regulates Alternative Splicing by Modulating SR Splicing Factor Phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Sci. 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Mohamed, J.S.; Gaughwin, P.M.; Lim, B.; Robson, P.; Lipovich, L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 2009, 16, 324–337. [Google Scholar] [CrossRef]
- A Espinoza, C.; A Allen, T.; Hieb, A.R.; Kugel, J.F.; A Goodrich, J. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat. Struct. Mol. Boil. 2004, 11, 822–829. [Google Scholar] [CrossRef]
- Shamovsky, I.; Ivannikov, M.; Kandel, E.S.; Gershon, D.; Nudler, E. RNA-mediated response to heat shock in mammalian cells. Nature 2006, 440, 556–560. [Google Scholar] [CrossRef]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.-H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Önder, T.T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Boil. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer — implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Miranda-Castro, R.; De-Los-Santos-Álvarez, N.; Lobo-Castañón, M.J. Long noncoding RNAs: from genomic junk to rising stars in the early detection of cancer. Anal. Bioanal. Chem. 2019, 411, 4265–4275. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.M.; Verjovski-Almeida, S. Perspectives of Long Non-Coding RNAs in Cancer Diagnostics. Front. Genet. 2012, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Campbell, M.E.; Goldberg, R.M.; Grothey, A.; Seitz, J.-F.; Benedetti, J.K.; André, T.; Haller, D.G.; Sargent, D.J. Survival Following Recurrence in Stage II and III Colon Cancer: Findings From the ACCENT Data Set. J. Clin. Oncol. 2008, 26, 2336–2341. [Google Scholar] [CrossRef]
- Lee, H.; Kim, C.; Ku, J.-L.; Kim, W.; Yoon, S.K.; Kuh, H.-J.; Lee, J.-H.; Nam, S.W.; Lee, E.K. A long non-coding RNA snaR contributes to 5-fluorouracil resistance in human colon cancer cells. Mol. Cells 2014, 37, 540–546. [Google Scholar] [CrossRef]
- Jiang, W.; Xia, J.; Xie, S.; Zou, R.; Pan, S.; Wang, P.; Assaraf, Y.G.; Zhu, X. Long non-coding RNAs as a determinant of cancer drug resistance: Towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updat. 2020, 50, 100683. [Google Scholar] [CrossRef]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2015, 44, D980–D985. [Google Scholar] [CrossRef]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.V.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.; Dinger, M.E. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2014, 43, D168–D173. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Liu, L.; Zhou, Y.; Zhang, S.; Chouvardas, P.; Johnson, R.; Li, J. LnCompare: gene set feature analysis for human long non-coding RNAs. Nucleic Acids Res. 2019, 47, W523–W529. [Google Scholar] [CrossRef]
- Fiorenzano, A.; Pascale, E.; Gagliardi, M.; Terreri, S.; Papa, M.; Andolfi, G.; Galasso, M.; Tagliazucchi, G.M.; Taccioli, C.; Patriarca, E.J.; et al. An Ultraconserved Element Containing lncRNA Preserves Transcriptional Dynamics and Maintains ESC Self-Renewal. Stem Cell Rep. 2018, 10, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zou, Y.; Hu, G.; Lin, C.; Guo, Y.; Gao, K.; Wu, M. Facilitating colorectal cancer cell metastasis by targeted binding of long non-coding RNA ENSG00000231881 with miR-133b via VEGFC signaling pathway. Biochem. Biophys. Res. Commun. 2019, 509, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-Y.; Wang, W.; Sun, J.-Y.; Xin, B.; Zhang, X.; Wang, T.; Zhang, Q.-F.; Yao, L.-B.; Han, H.; Fan, D.-M.; et al. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018, 8, 2846–2861. [Google Scholar] [CrossRef] [PubMed]
- Shioiri, M.; Shida, T.; Koda, K.; Seike, K.; Nishimura, M.; Takano, S.; Miyazaki, M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br. J. Cancer 2006, 94, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, J.; Lei, L.; He, H.; Chen, E.; Dong, J.; Yang, J. High Vimentin Expression Predicts a Poor Prognosis and Progression in Colorectal Cancer: A Study with Meta-Analysis and TCGA Database. BioMed Res. Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Goel, A.; Kusunoki, M. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinog. 2013, 34, 2548–2557. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest Cancer Res 2012, 5, 19–27. [Google Scholar]
- Minotti, L.; Agnoletto, C.; Baldassari, F.; Corrà, F.; Volinia, S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput 2018, 7, 34. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Lu, H.; Liu, H.; Yang, X.; Ye, T.; Lv, P.; Wu, X.; Ye, Z. LncRNA BLACAT1 May Serve as a Prognostic Predictor in Cancer: Evidence from a Meta-Analysis. BioMed Res. Int. 2019, 2019, 1275491. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wen, J.; Gao, P.; Zhang, G.; Zhang, G. Overexpression of the long non-coding RNA, linc-UBC1, is associated with poor prognosis and facilitates cell proliferation, migration, and invasion in colorectal cancer. OncoTargets Ther. 2017, 10, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hou, Z.; Zuo, S.; Zhou, X.; Feng, Y.; Sun, Y.; Yuan, X. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40- MDM 2-p53 pathway through binding with UBA 52. Cancer Sci. 2019, 110, 1194–1207. [Google Scholar] [CrossRef]
- Yang, L.; Qiu, M.; Xu, Y.; Wang, J.; Zheng, Y.; Li, M.; Xu, L.; Yin, R. Upregulation of long non-coding RNA PRNCR1 in colorectal cancer promotes cell proliferation and cell cycle progression. Oncol. Rep. 2015, 35, 318–324. [Google Scholar] [CrossRef]
- Zheng, Y.; Nie, P.; Xu, S. Long noncoding RNA CASC21 exerts an oncogenic role in colorectal cancer through regulating miR-7-5p/YAP1 axis. Biomed. Pharmacother. 2020, 121, 109628. [Google Scholar] [CrossRef] [PubMed]
- Idogawa, M.; Ohashi, T.; Sasaki, Y.; Nakase, H.; Tokino, T. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int. J. Cancer 2017, 140, 2785–2791. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res. 2011, 40, D84–D90. [Google Scholar] [CrossRef]
- Andrews, S.; Bittencourt, A.S. FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 January 2017).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; A Irizarry, R.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Brennan, K.; Hernaez, M.; Gevaert, O. Benchmark of long non-coding RNA quantification for RNA sequencing of cancer samples. GigaScience 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 002832. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. New Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Nitsche, A.; Rose, M.; Fasold, M.; Missal, K.; Stadler, P.F. Comparison of splice sites reveals that long noncoding RNAs are evolutionarily well conserved. RNA 2015, 21, 801–812. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.E.; Ye, Z.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.A.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahrour, F.; Díaz-Uriarte, R.; Dopazo, J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iraola-Guzmán, S.; Brunet-Vega, A.; Pegueroles, C.; Saus, E.; Hovhannisyan, H.; Casalots, A.; Pericay, C.; Gabaldón, T. Target Enrichment Enables the Discovery of lncRNAs with Somatic Mutations or Altered Expression in Paraffin-Embedded Colorectal Cancer Samples. Cancers 2020, 12, 2844. https://doi.org/10.3390/cancers12102844
Iraola-Guzmán S, Brunet-Vega A, Pegueroles C, Saus E, Hovhannisyan H, Casalots A, Pericay C, Gabaldón T. Target Enrichment Enables the Discovery of lncRNAs with Somatic Mutations or Altered Expression in Paraffin-Embedded Colorectal Cancer Samples. Cancers. 2020; 12(10):2844. https://doi.org/10.3390/cancers12102844
Chicago/Turabian StyleIraola-Guzmán, Susana, Anna Brunet-Vega, Cinta Pegueroles, Ester Saus, Hrant Hovhannisyan, Alex Casalots, Carles Pericay, and Toni Gabaldón. 2020. "Target Enrichment Enables the Discovery of lncRNAs with Somatic Mutations or Altered Expression in Paraffin-Embedded Colorectal Cancer Samples" Cancers 12, no. 10: 2844. https://doi.org/10.3390/cancers12102844
APA StyleIraola-Guzmán, S., Brunet-Vega, A., Pegueroles, C., Saus, E., Hovhannisyan, H., Casalots, A., Pericay, C., & Gabaldón, T. (2020). Target Enrichment Enables the Discovery of lncRNAs with Somatic Mutations or Altered Expression in Paraffin-Embedded Colorectal Cancer Samples. Cancers, 12(10), 2844. https://doi.org/10.3390/cancers12102844