Under-Replicated DNA: The Byproduct of Large Genomes?




{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Eukaryotic Genome Replication Challenges
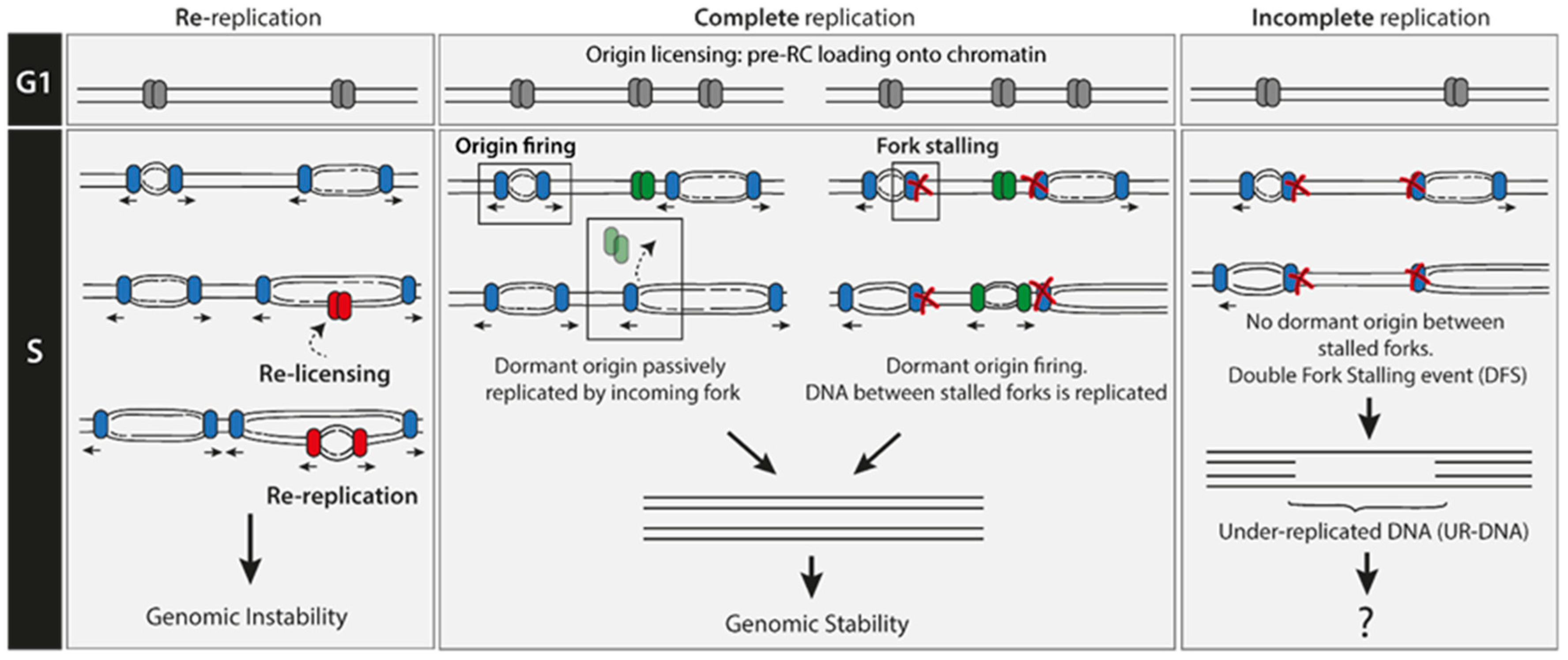
2. Re-Replication Events Are Rare
3. Under-Replication Events Are Frequent
3.1. Double Fork Stalling (DFS) Events as the Main Source of UR-DNA
3.2. DNA Loci Which Are Recurrently Prone to Suffer DFS
3.3. Faulty DNA Replication Termination as a Potential Source of UR-DNA
4. How Under-Replicated DNA Escapes Detection by the S-Phase Checkpoint?
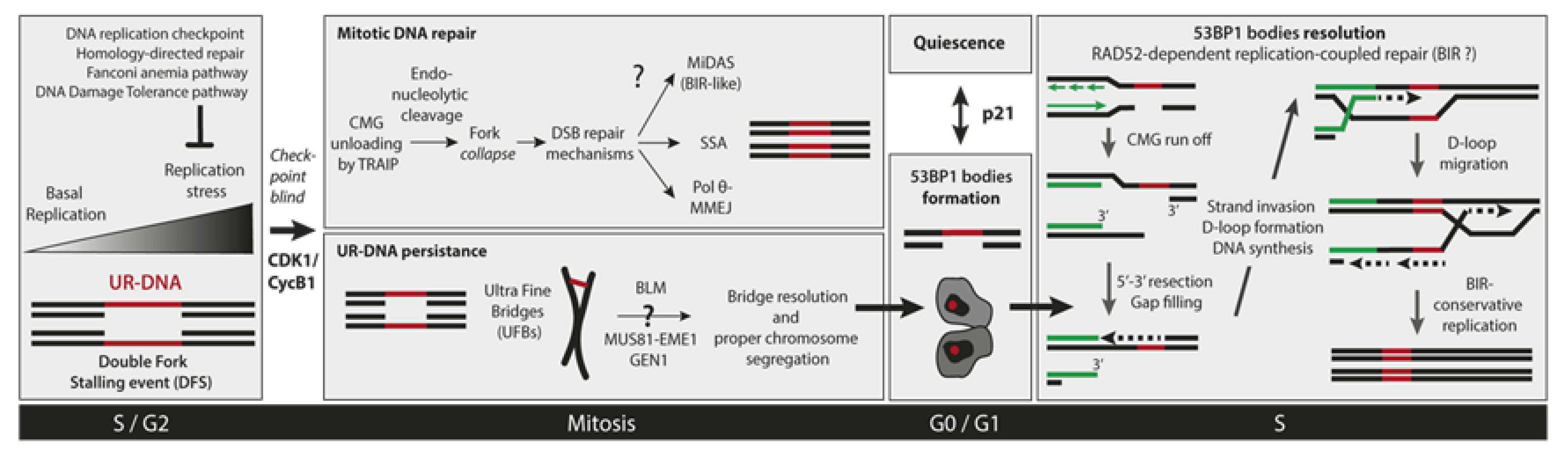
5. Cellular Consequences of Incomplete DNA Replication
6. Mitotic DNA Repair
6.1. Mitotic DNA Synthesis
6.2. Mitotic DNA End Joining Events
7. UR-DNA Segregation Defects: Ultra-Fine Bridges
8. 53BP1 Nuclear Bodies
8.1. 53BP1 NBs: Backup for Insufficient MiDAS or Primary Choice?
8.2. 53BP1-NBs Resolution
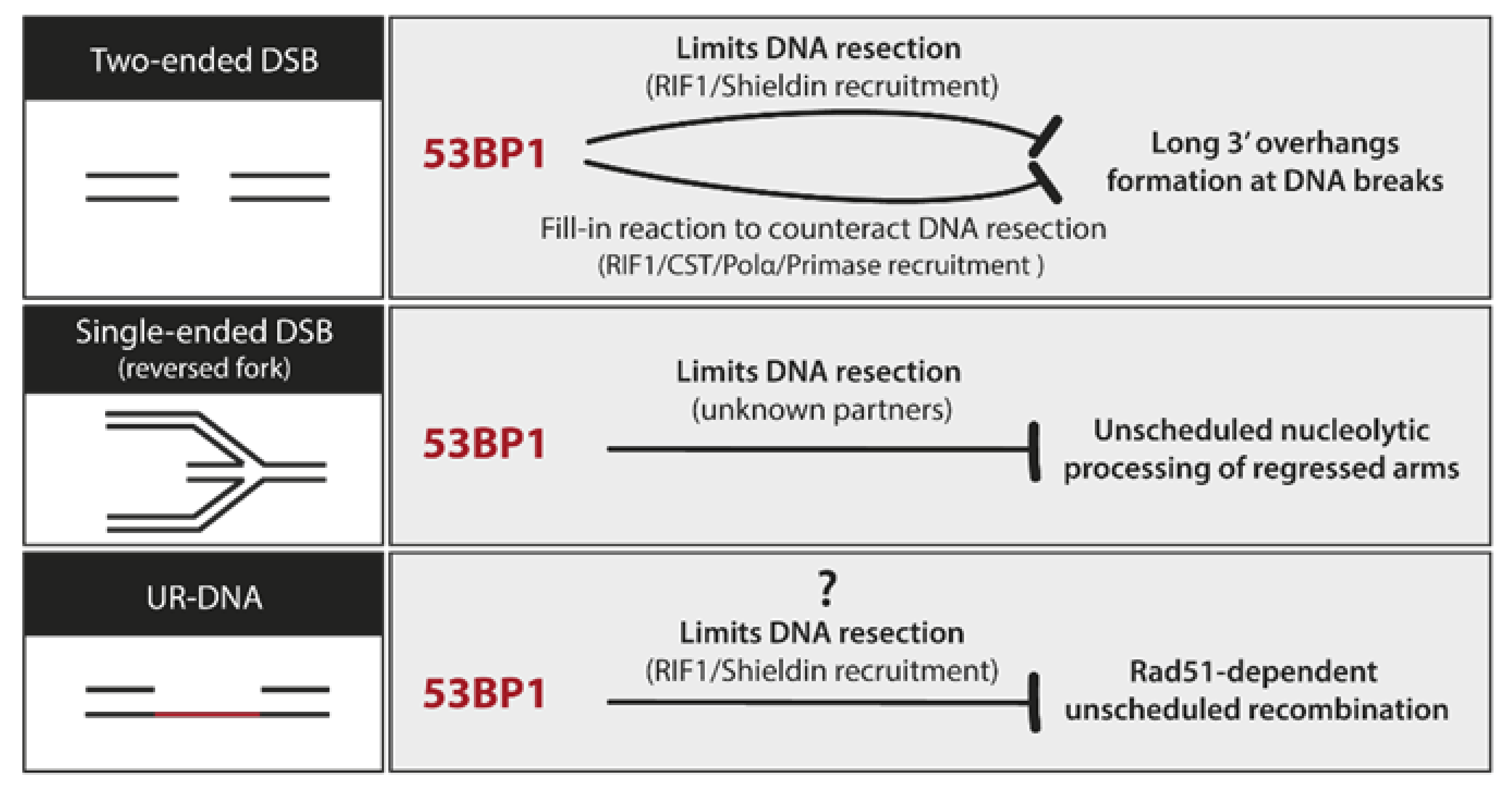
8.3. UR-DNA Safeguarding Properties of 53BP1
9. The Biological Significance of Transgenerational Transmission of DNA Damage
10. The Enigmatic Error-Prone Nature of UR-DNA Repair
11. Clinical Relevance and Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a010108. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F. Quality control in the initiation of eukaryotic DNA replication. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 3545–3553. [Google Scholar] [CrossRef]
- Davidson, I.F.; Li, A.; Blow, J.J. Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol. Cell 2006, 24, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Lee, A.Y.; Chiba, T.; Olson, E.; Sun, P.; Wu, X. The ATR-mediated S phase checkpoint prevents rereplication in mammalian cells when licensing control is disrupted. J. Cell Biol. 2007, 179, 643–657. [Google Scholar] [CrossRef]
- Green, B.M.; Finn, K.J.; Li, J.J. Loss of DNA replication control is a potent inducer of gene amplification. Science 2010, 329, 943–946. [Google Scholar] [CrossRef]
- Hanlon, S.L.; Li, J.J. Re-replication of a centromere induces chromosomal instability and aneuploidy. PLoS Genet. 2015, 11, e1005039. [Google Scholar] [CrossRef]
- Munoz, S.; Bua, S.; Rodriguez-Acebes, S.; Megias, D.; Ortega, S.; de Martino, A.; Mendez, J. In vivo DNA re-replication elicits lethal tissue dysplasias. Cell Rep. 2017, 19, 928–938. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Zanini, I.M.; Mijic, S.; Herrador, R.; Zellweger, R.; Chaudhuri, A.R.; Creavin, K.D.; Blow, J.J.; Lopes, M. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013, 27, 2537–2542. [Google Scholar] [CrossRef]
- Melixetian, M.; Ballabeni, A.; Masiero, L.; Gasparini, P.; Zamponi, R.; Bartek, J.; Lukas, J.; Helin, K. Loss of Geminin induces rereplication in the presence of functional p53. J. Cell Biol. 2004, 165, 473–482. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, Y.; Dutta, A. Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol. Cell. Biol. 2004, 24, 7140–7150. [Google Scholar] [CrossRef]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Duronio, R.J. Endoreplication and polyploidy: Insights into development and disease. Development 2013, 140, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nordman, J.; Orr-Weaver, T.L. Regulation of DNA replication during development. Development 2012, 139, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef]
- Diffley, J.F. Once and only once upon a time: Specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev. 1996, 10, 2819–2830. [Google Scholar] [CrossRef]
- Hook, S.S.; Lin, J.J.; Dutta, A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007, 19, 663–671. [Google Scholar] [CrossRef]
- Vaziri, C.; Saxena, S.; Jeon, Y.; Lee, C.; Murata, K.; Machida, Y.; Wagle, N.; Hwang, D.S.; Dutta, A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 2003, 11, 997–1008. [Google Scholar] [CrossRef]
- Tatsumi, Y.; Sugimoto, N.; Yugawa, T.; Narisawa-Saito, M.; Kiyono, T.; Fujita, M. Deregulation of Cdt1 induces chromosomal damage without rereplication and leads to chromosomal instability. J. Cell Sci. 2006, 119, 3128–3140. [Google Scholar] [CrossRef]
- Blow, J.J.; Gillespie, P.J. Replication licensing and cancer--a fatal entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009, 10, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Mamun, M.A.; Albergante, L.; Moreno, A.; Carrington, J.T.; Blow, J.J.; Newman, T.J. Inevitability and containment of replication errors for eukaryotic genome lengths spanning megabase to gigabase. Proc. Natl. Acad. Sci. USA 2016, 113, E5765–E5774. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Mamun, M.A.; Haagensen, E.J.; Komseli, E.S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.J.; Mamun, M.A.; Nieduszynski, C.A.; Blow, J.J. Replisome stall events have shaped the distribution of replication origins in the genomes of yeasts. Nucleic Acids Res. 2013, 41, 9705–9718. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Hua, B.L.; Orr-Weaver, T.L. DNA replication control during drosophila development: Insights into the onset of s phase, replication initiation, and fork progression. Genetics 2017, 207, 29–47. [Google Scholar] [CrossRef]
- Macheret, M.; Bhowmick, R.; Sobkowiak, K.; Padayachy, L.; Mailler, J.; Hickson, I.D.; Halazonetis, T.D. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 2020, 1–12. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Ji, F.; Liao, H.; Pan, S.; Ouyang, L.; Jia, F.; Fu, Z.; Zhang, F.; Geng, X.; Wang, X.; Li, T.; et al. Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res. 2020, 1–15. [Google Scholar] [CrossRef]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Debacker, K.; Kooy, R.F. Fragile sites and human disease. Hum. Mol. Genet. 2007, 16, R150–R158. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, W.; Spradling, A.C. Incomplete replication generates somatic DNA alterations within Drosophila polytene salivary gland cells. Genes Dev. 2014, 28, 1840–1855. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.S.; Kleckner, N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002, 297, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Tsantoulis, P.; Kotsinas, A.; Michalopoulos, I.; Townsend, P.; Gorgoulis, V.G. Are common fragile sites merely structural domains or highly organized “functional” units susceptible to oncogenic stress? Cell. Mol. Life Sci. 2014, 71, 4519–4544. [Google Scholar] [CrossRef]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef]
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef]
- Steinacher, R.; Osman, F.; Dalgaard, J.Z.; Lorenz, A.; Whitby, M.C. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 2012, 26, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Baxter, J.; Ortiz Bazan, M.A.; Yeeles, J.T.P.; Labib, K.P.M. Pif1-family helicases support fork convergence during DNA replication termination in eukaryotes. Mol. Cell 2019, 74, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Budzowska, M.; Walter, J.C. The mechanism of DNA replication termination in vertebrates. Nature 2015, 525, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Moreno, S.P.; Knebel, A.; Johnson, C.; Hastie, C.J.; Gartner, A.; Gambus, A.; Labib, K. CUL-2(LRR-1) and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol. 2017, 19, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Mukherjee, P.; Tatham, M.H.; Hay, R.; Labib, K. Ufd1-Npl4 recruit Cdc48 for disassembly of ubiquitylated CMG helicase at the end of chromosome replication. Cell Rep. 2017, 18, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346, 1253596. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Mukherjee, P.P.; Fujisawa, R.; Rivera, C.P.; Labib, K. CMG helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. Elife 2020, 9, 9. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A.; Lloyd, R.G. Avoiding chromosome pathology when replication forks collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef]
- Wendel, B.M.; Courcelle, C.T.; Courcelle, J. Completion of DNA replication in escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 16454–16459. [Google Scholar] [CrossRef]
- Wendel, B.M.; Cole, J.M.; Courcelle, C.T.; Courcelle, J. SbcC-SbcD and ExoI process convergent forks to complete chromosome replication. Proc. Natl. Acad. Sci. USA 2018, 115, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Wendel, B.M.; Livingstone, D.D.; Courcelle, C.T. RecBCD is required to complete chromosomal replication: Implications for double-strand break frequencies and repair mechanisms. DNA Repair 2015, 32, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; Possoz, C.; Durand, A.; Desfontaines, J.M.; Barre, F.X.; Leach, D.R.F.; Michel, B. Broken replication forks trigger heritable DNA breaks in the terminus of a circular chromosome. PLoS Genet. 2018, 14, e1007256. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.A.; Wendel, B.M.; Weber, E.A.; Courcelle, C.T.; Courcelle, J. RecBCD, SbcCD and ExoI process a substrate created by convergent replisomes to complete DNA replication. Mol. Microbiol. 2019, 111, 1638–1651. [Google Scholar] [CrossRef]
- Dimude, J.U.; Midgley-Smith, S.L.; Stein, M.; Rudolph, C.J. Replication termination: Containing fork fusion-mediated pathologies in escherichia coli. Genes 2016, 7, 40. [Google Scholar] [CrossRef]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; De Piccoli, G.; Cordon-Preciado, V.; Farmer, S.; Jarmuz, A.; Machin, F.; Pasero, P.; Lisby, M.; Haber, J.E.; Aragon, L. Anaphase onset before complete DNA replication with intact checkpoint responses. Science 2007, 315, 1411–1415. [Google Scholar] [CrossRef]
- Lobrich, M.; Jeggo, P.A. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 2007, 7, 861–869. [Google Scholar] [CrossRef]
- van den Berg, J.; Manjón, A.G.; Kielbassa, K.; Feringa, F.M.; Freire, R.; Medema, R.H. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018, 46, 10132–10144. [Google Scholar] [CrossRef]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 2018, 71, 117–128. [Google Scholar] [CrossRef]
- Lemmens, B.; Lindqvist, A. DNA replication and mitotic entry: A brake model for cell cycle progression. J. Cell Biol. 2019, 218, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Techer, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Marco, S.D.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Huhn, D.; et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 2017, 66, 658–671.e8. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Wu, W.; Bhowmick, R.; Vogel, I.; Ozer, O.; Ghisays, F.; Thakur, R.S.; De Leon, E.S.; Richter, P.H.; Ren, L.; Petrini, J.H.; et al. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020, 27, 424–437. [Google Scholar] [CrossRef]
- Priego Moreno, S.; Jones, R.M.; Poovathumkadavil, D.; Scaramuzza, S.; Gambus, A. Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK promotes replisome disassembly, fork breakage, and complex DNA rearrangements. Mol. Cell 2019, 73, 915–929.e6. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Replication-coupled DNA repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef]
- Downing, B.; Morgan, R.; VanHulle, K.; Deem, A.; Malkova, A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat. Res. 2008, 645, 9–18. [Google Scholar] [CrossRef]
- Wilson, M.A.; Kwon, Y.; Xu, Y.; Chung, W.H.; Chi, P.; Niu, H.; Mayle, R.; Chen, X.; Malkova, A.; Sung, P.; et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 2013, 502, 393–396. [Google Scholar] [CrossRef]
- Garribba, L.; Bjerregaard, V.A.; Dinis, M.M.G.; Ozer, O.; Wu, W.; Sakellariou, D.; Pena-Diaz, J.; Hickson, I.D.; Liu, Y. Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16527–16536. [Google Scholar] [CrossRef]
- Graber-Feesl, C.L.; Pederson, K.D.; Aney, K.J.; Shima, N. Mitotic DNA synthesis is differentially regulated between cancer and noncancerous cells. Mol. Cancer Res. 2019, 17, 1687–1698. [Google Scholar] [CrossRef]
- Duda, H.; Arter, M.; Gloggnitzer, J.; Teloni, F.; Wild, P.; Blanco, M.G.; Altmeyer, M.; Matos, J. A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev. Cell 2016, 39, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Bizard, A.H.; Hickson, I.D. Anaphase: A fortune-teller of genomic instability. Curr. Opin. Cell Biol. 2018, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.; Korner, R.; Hofmann, K.; Nigg, E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007, 128, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef]
- Sarlos, K.; Biebricher, A.S.; Bizard, A.H.; Bakx, J.A.M.; Ferrete-Bonastre, A.G.; Modesti, M.; Paramasivam, M.; Yao, Q.; Peterman, E.J.G.; Wuite, G.J.L.; et al. Reconstitution of anaphase DNA bridge recognition and disjunction. Nat. Struct. Mol. Biol. 2018, 25, 868–876. [Google Scholar] [CrossRef]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. p53 Binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Jasin, M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Luebben, S.W.; Kawabata, T.; Johnson, C.S.; O’Sullivan, M.G.; Shima, N. A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res. 2014, 42, 5605–5615. [Google Scholar] [CrossRef]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef]
- Dave, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep. 2014, 7, 53–61. [Google Scholar] [CrossRef]
- Hiraga, S.; Alvino, G.M.; Chang, F.; Lian, H.Y.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.K.; Donaldson, A.D. Rif1 controls DNA replication by directing protein phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Iwabuchi, K.; Bartel, P.L.; Li, B.; Marraccino, R.; Fields, S. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA 1994, 91, 6098–6102. [Google Scholar] [CrossRef]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Morris, J.R. Moving mountains-the BRCA1 promotion of DNA resection. Front. Mol. Biosci. 2019, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Schmid, J.A.; Berti, M.; Walser, F.; Raso, M.C.; Schmid, F.; Krietsch, J.; Stoy, H.; Zwicky, K.; Ursich, S.; Freire, R.; et al. Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol. Cell 2018, 71, 897–910.e8. [Google Scholar] [CrossRef]
- Her, J.; Ray, C.; Altshuler, J.; Zheng, H.; Bunting, S.F. 53BP1 mediates ATR-Chk1 signaling and protects replication forks under conditions of replication stress. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Arora, M.; Moser, J.; Phadke, H.; Basha, A.A.; Spencer, S.L. Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 2017, 19, 1351–1364. [Google Scholar] [CrossRef]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novak, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef] [PubMed]
- Lezaja, A.; Altmeyer, M. Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle 2018, 17, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Donnianni, R.A.; Symington, L.S. Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 13475–13480. [Google Scholar] [CrossRef]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Pogliano, C.B.; Matos, J.; et al. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-transcriptional r-loops. Mol. Cell 2020, 77, 528–541.e8. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative lengthening of telomeres mediated by mitotic DNA synthesis engages break-induced replication processes. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Mayle, R.; Campbell, I.M.; Beck, C.R.; Yu, Y.; Wilson, M.; Shaw, C.A.; Bjergbaek, L.; Lupski, J.R.; Ira, G. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 2015, 349, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E deregulation promotes loss of specific genomic regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA damaging revolution: PARP inhibitors and beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertolin, A.P.; Hoffmann, J.-S.; Gottifredi, V. Under-Replicated DNA: The Byproduct of Large Genomes? Cancers 2020, 12, 2764. https://doi.org/10.3390/cancers12102764
Bertolin AP, Hoffmann J-S, Gottifredi V. Under-Replicated DNA: The Byproduct of Large Genomes? Cancers. 2020; 12(10):2764. https://doi.org/10.3390/cancers12102764
Chicago/Turabian StyleBertolin, Agustina P., Jean-Sébastien Hoffmann, and Vanesa Gottifredi. 2020. "Under-Replicated DNA: The Byproduct of Large Genomes?" Cancers 12, no. 10: 2764. https://doi.org/10.3390/cancers12102764
APA StyleBertolin, A. P., Hoffmann, J.-S., & Gottifredi, V. (2020). Under-Replicated DNA: The Byproduct of Large Genomes? Cancers, 12(10), 2764. https://doi.org/10.3390/cancers12102764