AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. AXL is Overexpressed in Mesothelioma, and Correlated with Poor Survival
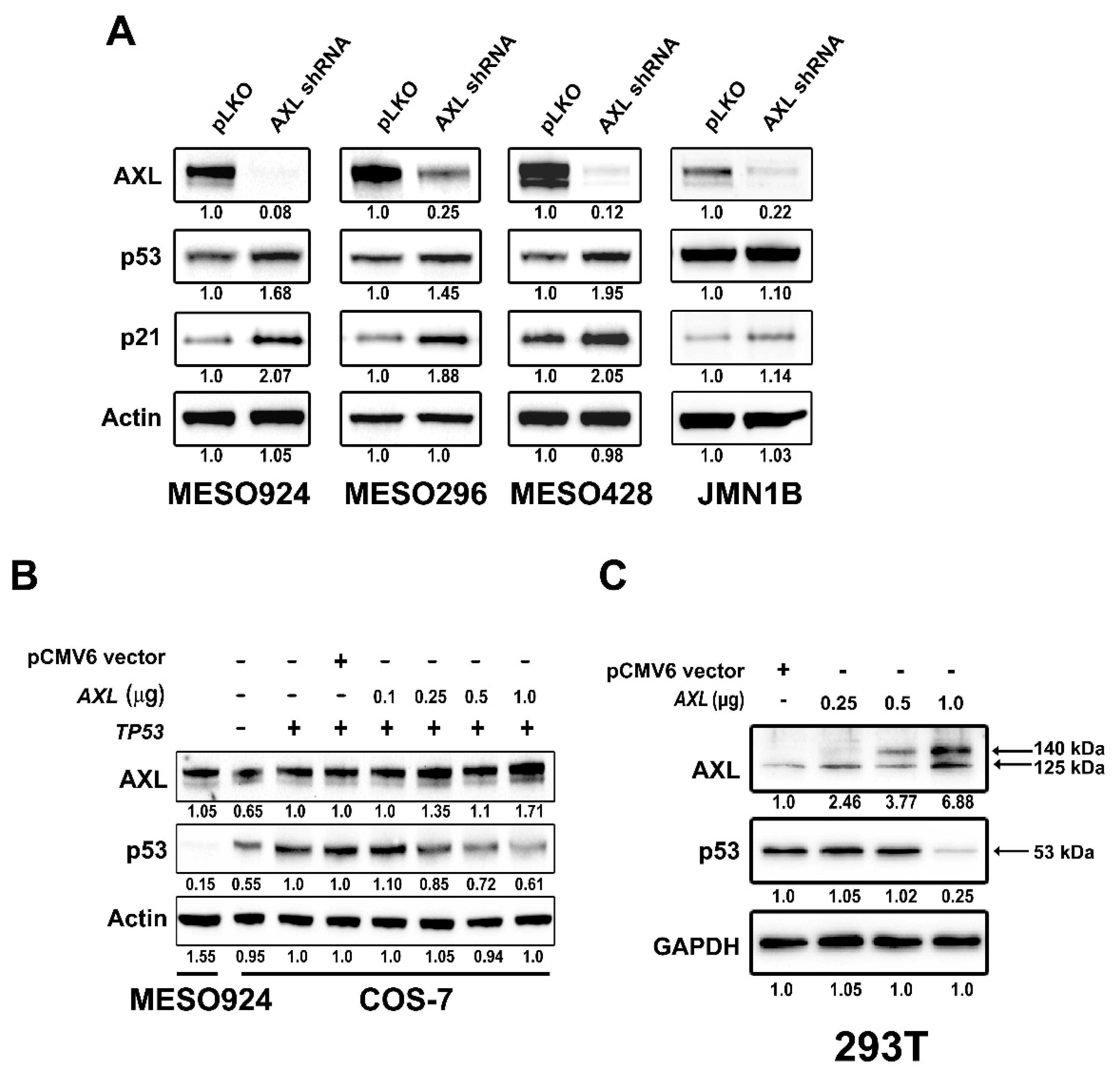
2.2. AXL Regulates p53 Protein Expression in Mesothelioma
2.3. No Protein Interaction is Detected between AXL and p53
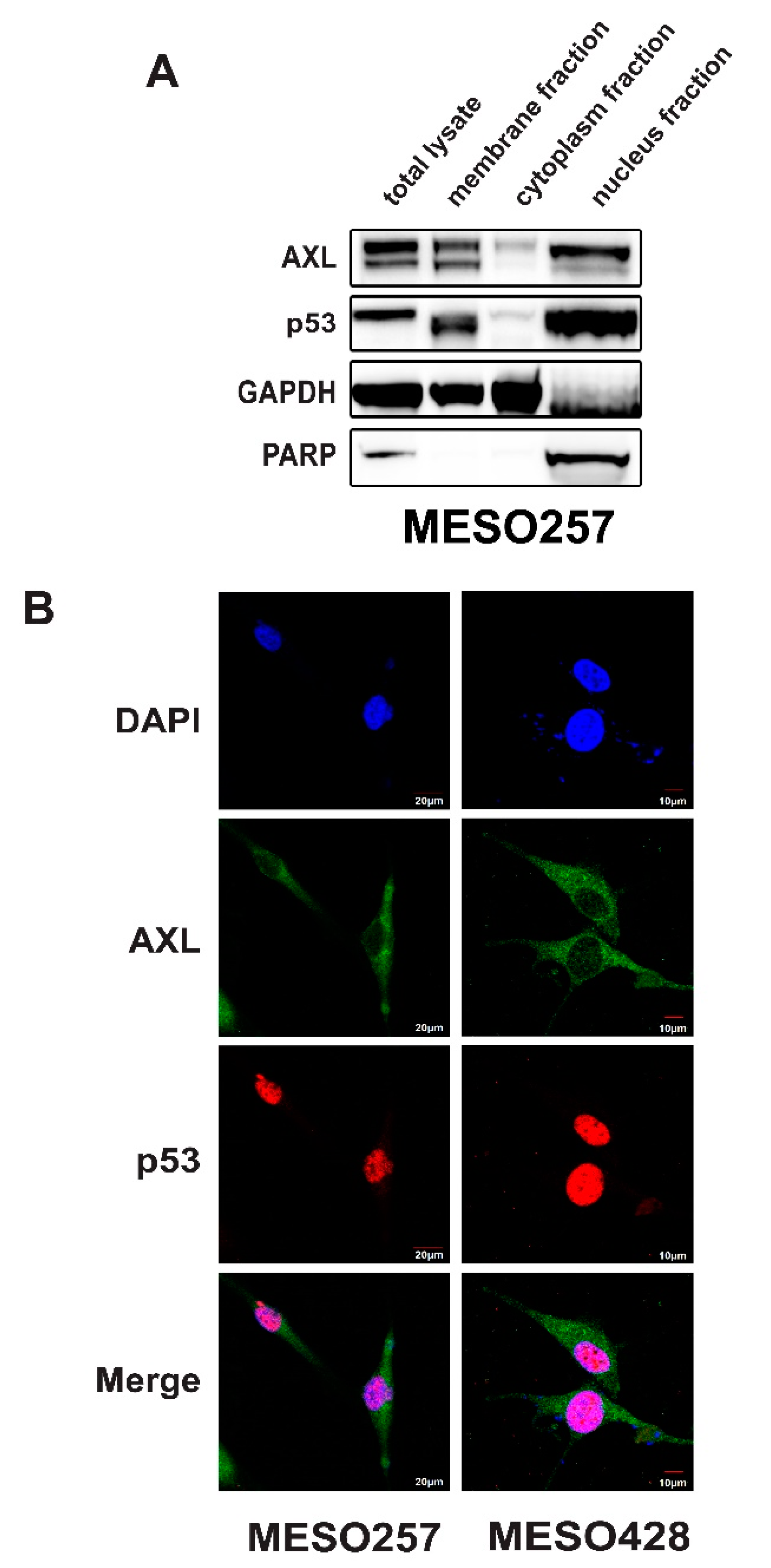
2.4. AXL Colocalization with p53 in the Nucleus
2.5. AXL Negatively Regulates TP53 Transcription in Mesothelioma
2.6. AXL Regulates Mesothelioma Migration, Invasiveness, and Proliferation through p53
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Mesothelioma Cell Lines and Frozen Tumor Specimens
4.3. Immunoblotting
4.4. Immunoprecipitation
4.5. Lentiviral AXL and p53 shRNA Constructs
4.6. Cell Culture and Virus Infection
4.7. Transfection
4.8. RNA Preparation and qRT-PCR
4.9. Immunofluorescence Staining
4.10. Dual Luciferase Assay
4.11. Chromatin Immunoprecipitation Assay (ChIP)
4.12. Cell Viability Analysis
4.13. In Vitro Wound Healing Assays
4.14. Cell Invasion Assays
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. WHO Classification of Tumors of the Lung, Pleura, Thymus and Heart; IARC Press: Lyon, France, 2015. [Google Scholar]
- Carbone, M.; Adusumilli, P.S.; Alexander, H.R.; Baas, P., Jr.; Bardelli, F.; Bononi, A.; Bueno, R.; Felley-Bosco, E.; Galateau-Salle, F.; Jablons, D.; et al. Mesothelioma: Scientific clues for prevention, diagnosis, and therapy. CA Cancer J. Clin. 2019, 69, 402–429. [Google Scholar] [CrossRef]
- Chatwal, M.S.; Tanvetyanon, T. Malignant mesothelioma clinical trial combines immunotherapy drugs. Immunotherapy 2018, 10, 341–344. [Google Scholar] [CrossRef]
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630. [Google Scholar] [CrossRef]
- Metaxas, Y.; Rivalland, G.; Mauti, L.A.; Klingbiel, D.; Kao, S.; Schmid, S.; Nowak, A.K.; Gautschi, O.; Bartnick, T.; Hughes, B.G.; et al. Pembrolizumab as Palliative Immunotherapy in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 1784–1791. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Zauderer, M.G. Nivo-lution in Mesothelioma. Clin. Cancer Res. 2019, 25, 5438–5440. [Google Scholar] [CrossRef]
- Pasello, G.; Zago, G.; Lunardi, F.; Urso, L.; Kern, I.; Vlacic, G.; Grosso, F.; Mencoboni, M.; Ceresoli, G.L.; Schiavon, M.; et al. Malignant pleural mesothelioma immune microenvironment and checkpoint expression: Correlation with clinical-pathological features and intratumor heterogeneity over time. Ann. Oncol. 2018, 29, 1258–1265. [Google Scholar] [CrossRef]
- Rosen, L.E.; Karrison, T.; Ananthanarayanan, V.; Gallan, A.J.; Adusumilli, P.S.; Alchami, F.S.; Attanoos, R.; Brcic, L.; Butnor, K.J.; Galateau-Sallé, F.; et al. Nuclear grade and necrosis predict prognosis in malignant epithelioid pleural mesothelioma: A multi-institutional study. Mod. Pathol. 2018, 31, 598–606. [Google Scholar] [CrossRef]
- Pelosi, G.; Papotti, M.; Righi, L.; Rossi, G.; Ferrero, S.; Bosari, S.; Calabrese, F.; Kern, I.; Maisonneuve, P.; Sonzogni, A.; et al. Pathologic Grading of Malignant Pleural Mesothelioma: An Evidence-Based Proposal. J. Thorac. Oncol. 2018, 13, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Serio, G.; Fortarezza, F.; Scattone, A.; Caporusso, C.; Punzi, A.; Cavone, D.; Pennella, A.; Marzullo, A.; Vimercati, L. Prognostic Value of Ki67 Percentage, WT-1 Expression and p16/CDKN2A Deletion in Diffuse Malignant Peritoneal Mesothelioma: A Single-Centre Cohort Study. Diagnostics 2020, 10, 386. [Google Scholar] [CrossRef]
- Ou, W.B.; Corson, J.M.; Flynn, D.L.; Lu, W.P.; Wise, S.C.; Bueno, R.; Sugarbaker, D.J.; Fletcher, J.A. AXL regulates mesothelioma proliferation and invasiveness. Oncogene 2011, 30, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Hubert, C.; Corson, J.M.; Bueno, R.; Flynn, D.L.; Sugarbaker, D.J.; Fletcher, J.A. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia 2011, 13, 12–22. [Google Scholar] [CrossRef]
- Husain, H.; Scur, M.; Murtuza, A.; Bui, N.; Woodward, B.; Kurzrock, R. Strategies to Overcome Bypass Mechanisms Mediating Clinical Resistance to EGFR Tyrosine Kinase Inhibition in Lung Cancer. Mol. Cancer Ther. 2017, 16, 265–272. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef]
- Ruprecht, B.; Zaal, E.A.; Zecha, J.; Wu, W.; Berkers, C.R.; Kuster, B.; Lemeer, S. Lapatinib Resistance in Breast Cancer Cells Is Accompanied by Phosphorylation-Mediated Reprogramming of Glycolysis. Cancer Res. 2017, 77, 1842–1853. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Okubo, T.; Saito, T.; Mitomi, H.; Takagi, T.; Torigoe, T.; Suehara, Y.; Kaneko, K.; Yao, T. p53 mutations may be involved in malignant transformation of giant cell tumor of bone through interaction with GPX1. Virchows Arch. 2013, 463, 67–77. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Vaughan, C.A.; Singh, S.; Windle, B.; Yeudall, W.A.; Frum, R.; Grossman, S.R.; Deb, S.P.; Deb, S. Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer 2012, 3, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Boysen, J.; Sinha, S.; Price-Troska, T.; Warner, S.L.; Bearss, D.J.; Viswanatha, D.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: Implications for therapy in p53-defective CLL patients. Leukemia 2014, 28, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, W.M.; Al-Khayyal, N.A.; Nair, V.A.; Aravind, S.R.; Saber-Ayad, M. Role of AXL in invasion and drug resistance of colon and breast cancer cells and its association with p53 alterations. World J. Gastroenterol. 2017, 23, 3440–3448. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Lu, M.; Eilers, G.; Li, H.; Ding, J.; Meng, X.; Wu, Y.; He, Q.; Sheng, Q.; Zhou, H.M.; et al. Co-targeting of FAK and MDM2 triggers additive anti-proliferative effects in mesothelioma via a coordinated reactivation of p53. Br. J. Cancer 2016, 115, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Hopkins-Donaldson, S.; Belyanskaya, L.L.; Simoes-Wust, A.P.; Sigrist, B.; Kurtz, S.; Zangemeister-Wittke, U.; Stahel, R. p53-induced apoptosis occurs in the absence of p14(ARF) in malignant pleural mesothelioma. Neoplasia 2006, 8, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Zuo, R.; Ni, N.; Eilers, G.; Wu, D.; Pei, Y.; Nie, Z.; Wu, Y.; Wu, Y.; Ou, W.B. Activated tyrosine kinases in gastrointestinal stromal tumor with loss of KIT oncoprotein expression. Cell Cycle 2018, 17, 2577–2592. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, L.; Li, H.; Eilers, G.; Kuang, Y.; Shi, S.; Yan, Z.; Li, X.; Corson, J.M.; Meng, F.; et al. Multipoint targeting of the PI3K/mTOR pathway in mesothelioma. Br. J. Cancer 2014, 110, 2479–2488. [Google Scholar] [CrossRef]
- Metcalf, R.A.; Welsh, J.A.; Bennett, W.P.; Seddon, M.B.; Lehman, T.A.; Pelin, K.; Linnainmaa, K.; Tammilehto, L.; Mattson, K.; Gerwin, B.I. p53 and Kirsten-ras mutations in human mesothelioma cell lines. Cancer Res. 1992, 52, 2610–2615. [Google Scholar]
- Yang, P.; Chen, W.; Li, X.; Eilers, G.; He, Q.; Liu, L.; Wu, Y.; Wu, Y.; Yu, W.; Fletcher, J.A.; et al. Downregulation of cyclin D1 sensitizes cancer cells to MDM2 antagonist Nutlin-3. Oncotarget 2016, 7, 32652–32663. [Google Scholar] [CrossRef]
- Ou, W.B.; Zhu, J.; Eilers, G.; Li, X.; Kuang, Y.; Liu, L.; Marino-Enriquez, A.; Yan, Z.; Li, H.; Meng, F.; et al. HDACi inhibits liposarcoma via targeting of the MDM2-p53 signaling axis and PTEN, irrespective of p53 mutational status. Oncotarget 2015, 6, 10510–10520. [Google Scholar] [CrossRef]
- Chen, M.K.; Hung, M.C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015, 282, 3693–3721. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Lien, H.C.; Xia, W.; Chen, I.F.; Lo, H.W.; Wang, Z.; Ali-Seyed, M.; Lee, D.F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Ems, S.M.; Lee, Y.W.; Stachowiak, E.K.; Pudavar, H.; Claus, P.; Prasad, P.N.; Stachowiak, M.K. Fibroblast growth factor receptor-1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Mol. Biol. Cell 2009, 20, 2401–2412. [Google Scholar] [CrossRef]
- Hsueh, Y.S.; Chang, H.H.; Shan, Y.S.; Sun, H.S.; Fletcher, J.A.; Li, C.F.; Chen, L.T. Nuclear KIT induces a NFKBIB-RELA-KIT autoregulatory loop in imatinib-resistant gastrointestinal stromal tumors. Oncogene 2019, 38, 6550–6565. [Google Scholar] [CrossRef]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017, 31, 1382–1397. [Google Scholar] [CrossRef]
- de Polo, A.; Luo, Z.; Gerarduzzi, C.; Chen, X.; Little, J.B.; Yuan, Z.M. AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J. Mol. Cell. Biol. 2017, 9, 154–165. [Google Scholar] [CrossRef]
- Ou, W.B.; Ni, N.; Zuo, R.; Zhuang, W.; Zhu, M.; Kyriazoglou, A.; Wu, D.; Eilers, G.; Demetri, G.D.; Qiu, H.; et al. Cyclin D1 is a mediator of gastrointestinal stromal tumor KIT-independence. Oncogene 2019, 38, 6615–6629. [Google Scholar] [CrossRef]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef]
- Raman, V.; Martensen, S.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Demetri, G.D.; Zenzie, B.W.; Rheinwald, J.G.; Griffin, J.D. Expression of colony-stimulating factor genes by normal human mesothelial cells and human malignant mesothelioma cells lines in vitro. Blood 1989, 74, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A.; et al. KIT Activation Is a Ubiquitous Feature of Gastrointestinal Stromal Tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar] [PubMed]
- Jiao, Y.; Ou, W.; Meng, F.; Zhou, H.; Wang, A. Targeting HSP90 in ovarian cancers with multiple receptor tyrosine kinase coactivation. Mol. Cancer 2011, 10, 125. [Google Scholar] [CrossRef]
- Malard, V.; Berenguer, F.; Prat, O.; Ruat, S.; Steinmetz, G.; Quemeneur, E. Global gene expression profiling in human lung cells exposed to cobalt. BMC Genomics 2007, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Thomazy, V.A.; Luthra, R.; Uthman, M.O.; Davies, P.J.; Medeiros, L.J. Determination of cyclin D1 and CD20 mRNA levels by real-time quantitative RT-PCR from archival tissue sections of mantle cell lymphoma and other non-Hodgkin’s lymphomas. J. Mol. Diagn. 2002, 4, 201–208. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Wang, H.; Lu, M.; Ni, X.; Bahri, N.; Zhu, S.; Chen, L.; Wu, Y.; Qiu, J.; Fletcher, J.A.; et al. AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression. Cancers 2020, 12, 2757. https://doi.org/10.3390/cancers12102757
Song W, Wang H, Lu M, Ni X, Bahri N, Zhu S, Chen L, Wu Y, Qiu J, Fletcher JA, et al. AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression. Cancers. 2020; 12(10):2757. https://doi.org/10.3390/cancers12102757
Chicago/Turabian StyleSong, Wei, Hao Wang, Minmin Lu, Xinxin Ni, Nacef Bahri, Shuihao Zhu, Limin Chen, Yuehong Wu, Jieqiong Qiu, Jonathan A. Fletcher, and et al. 2020. "AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression" Cancers 12, no. 10: 2757. https://doi.org/10.3390/cancers12102757
APA StyleSong, W., Wang, H., Lu, M., Ni, X., Bahri, N., Zhu, S., Chen, L., Wu, Y., Qiu, J., Fletcher, J. A., & Ou, W.-B. (2020). AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression. Cancers, 12(10), 2757. https://doi.org/10.3390/cancers12102757