1. Introduction
Sarcomas arising from soft tissues throughout the body, such as muscle, fat, and nerves, exhibit a variety of histological types, depending on their tissue of origin and degree of differentiation; their incidence is very low compared to solid tumors of epithelial tissue origin [
1,
2,
3]. Surgery is the primary option for the treatment of soft tissue sarcomas (STSs), with high survival rates expected after extensive resection. However, highly invasive surgical resection leads to poor postoperative quality of life and has a significant impact on patients’ physical, mental, and career development. Of note, children and young adults have a high incidence of STSs. Furthermore, due to the high hematogenous metastatic potential of sarcoma cells, patients with STSs are forced to live with the fear of recurrence and the appearance of distant metastasis, even if the primary tumor is resected [
4]. Importantly, because of the lack of effective drugs for patients manifesting recurrence/metastasis and for patients with non-surgically resectable STSs, the development of new therapeutics that can meet their medical needs is desperately required.
In recent years, novel cancer treatments such as immune checkpoint inhibitors and T-cell adoptive immunotherapy have demonstrated drastic therapeutic benefits for epithelial solid tumors [
5,
6,
7]. Although it is desirable to expand the application of these immunotherapies to STSs, their effectiveness is only limited to some, such as undifferentiated pleomorphic sarcoma, alveolar soft part sarcoma, and synovial sarcoma [
8,
9,
10,
11]. This is not only due to the fact that most STSs are cold tumors lacking tumor-associated antigens, which are the main targets of anti-cancer immune cells, but also because generally the tumor tissues are not infiltrated with anti-cancer immune cells [
12]. On the other hand, as in epithelial carcinomas, STSs induce angiogenesis from nearby parent vessels as the tumor grows, and vascular endothelial cells comprising these tumor vessels have been shown to strongly express vascular endothelial growth factor receptor 2 (VEGFR2) [
13,
14]. Previously, we have developed adoptive immunotherapy using T cells expressing a chimeric antigen receptor (CAR) that targets VEGFR2 and reported that anti-VEGFR2 CAR-T cells could inhibit tumor growth based on tumor vascular injury in a variety of tumor-bearing mouse models [
15,
16]. Our TACTICs (Tumor Angiogenesis-specific CAR-T cells Impacting Cancers) therapeutic strategy has the potential to be an all-around effective treatment against diverse STSs and to prevent hematogenous metastases via vascular injury. In fact, since it has been reported that cancer cells from some cases of angiosarcoma and synovial sarcoma express VEGFR2 [
14,
17], TACTICs therapy might demonstrate excellent tumor elimination by direct cytotoxicity towards the sarcoma cells. Therefore, we considered conducting a first in humans (FIH) study to evaluate the safety and efficacy of TACTICs therapy as a new treatment option for refractory STSs.
In every FIH study, ensuring the safety of the subjects is of the highest priority. CAR-T cells can be produced by gene transfer using viral vectors and provide long-term antitumor effects with a single dose; however, their use carries the risk of genotoxicity. Therefore, the safety management of subjects should be carried out continuously and consistently, as the complete elimination of transferred CAR-T cells from the body can be difficult when signs of serious adverse effects are observed [
18]. Therefore, we selected electroporation (EP) transfer of mRNA encoding the CAR gene as a method for generating CAR-T cells for clinical research [
16]. The mRNA-EP method can reduce the risk of side effects by allowing the reversion of CAR-T cells to their original phenotype over time; however, in this context, the efficacy of CAR-T cells is obviously transient. A previous study has confirmed that the mRNA-EP method was an efficient way to express CAR on T cells without affecting their viability and functions. Further, the study revealed that multiple doses of anti-VEGFR2 CAR-mRNA-transfected T cells exhibited strong anti-tumor activity, comparable to that provided by a single dose of CAR-T cells generated via viral vector-mediated gene transfer [
16].
In the preparation for the FIH study using mRNA-transfected CAR-T cells, we needed to determine the final development candidate (the specific anti-VEGFR2 CAR-T cells batch) and establish a method for producing and transporting 10
6–10
10 CAR-T cells to be administered in a single dose. In particular, the mRNA construct optimization for T cells’ transfection, to ensure a favorable CAR expression profile on their membranes, may improve the intensity and duration of CAR-T cells’ function [
19,
20]. In addition, removal of non-self-sequences in the CAR extracellular region as much as possible is expected to reduce the antigenicity of CAR-T cells and diminish their potential elimination by the host immune cells [
21]. Importantly, to properly evaluate the efficacy and safety of TACTICs therapy in a small population, it is necessary to select patients who are expected to respond well to this therapy. While, as abovementioned, angiosarcoma, synovial sarcoma, liposarcoma, and undifferentiated polymorphic sarcoma have been reported to strongly express VEGFR2, and are expected to be excellent targets for TACTICs therapy, not all STS cases are VEGFR2 positive [
13,
14]. Therefore, the development of companion diagnostics and the establishment of biomarkers are desirable to select VEGFR2-positive cases, potentially good responders to our anti-VEGFR2 CAR-T cell-based therapy.
In this study, we produced anti-VEGFR2 CAR-T cells for clinical research using a GMP-compatible EP-enabled electroporator. We optimized the CAR-mRNA construct to obtain the final development candidate for the anti-VEGFR2 CAR-T cell-based therapy. In addition, we selected the optimal transport conditions for mRNA-transfected CAR-T cells, from the cell manufacturing facility to the centers where STS patients are expected to be treated, in a way to maintain CAR-T cell functions. In addition, we collected information that allowed the selection of STS cases suitable for the future application of our TACTICs therapy: we predicted the potential efficacy and safety of our approach using immunohistochemical analysis of human STSs-derived and normal tissues.
3. Discussion
In this study, we first examined the specifications and transport conditions for the final development of a promising anti-VEGFR2 CAR-T cells candidate, in preparation for a future FIH study using TACTICs therapy. CAR-T cells generated via viral vectors are expected to have long-term anti-tumor effects due to persistent CAR expression; of note, CD137-derived STD are often incorporated into the second-generation CAR constructs to improve the in vivo persistence of CAR-T cells [
22,
23]. On the other hand, in CAR-T cells generated via mRNA-EP, memory differentiation and long-term survival of the transfected T cell are not as important, since they lose their CAR-mediated antigen specificity with time. In this study, we followed the second approach. In order to clearly support the mRNA-EP-derived CAR-T cells’ therapeutic potential (considering frequent administrations), it is essential to show that mRNA-transfected CAR-T cells exert a strong cytotoxic activity, at least in a short period of time. Indeed, here we determined that a second-generation CAR construct with a CD28-derived STD was suitable for producing our anti-VEGFR2 CAR-T cells. Of note, the nonspecific cytotoxic activity of CAR [V/8a/8a/137-3z]-T cells as shown in
Supplementary Figure S1D suggested that the second-generation CAR construct incorporating CD137-derived STD might have a high risk of causing off-target toxicity. Therefore, the design of CAR constructs targeting tumor-associated antigens which are also slightly expressed in normal tissues requires further validation regardless of the method of CAR-T cells production.
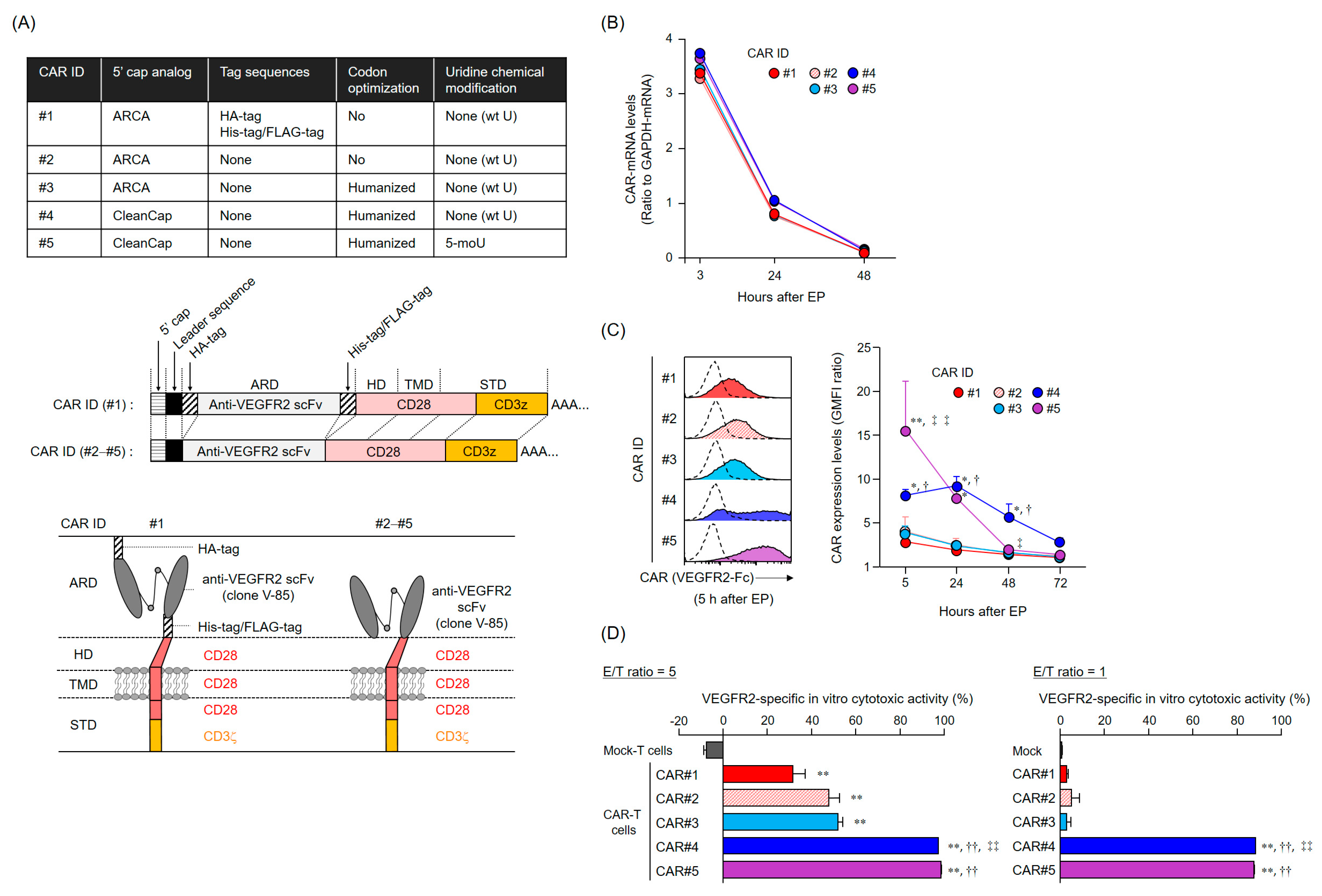
The anti-tumor activity of mRNA-transfected CAR-T cells depends on the CAR expression levels and duration. Importantly, CAR expression profiles are supposedly determined by the persistence of CAR-mRNA and its translation efficiency (rate). The five CAR-mRNAs examined in this study showed a similar mRNA elimination profile in the T cells, suggesting that the difference in the cell surface expression profiles of CAR#1–#5 was attributed to the amount and rate of synthesis of CAR proteins (translation efficiency). The ribosomes initiate translation via recognition of the cap structure at the 5′ end of mRNA. CleanCap is known to have a higher capping efficiency to in vitro transcribed RNA than ARCA [
19]. Therefore, we believed that the improvement in CAR expression levels due to the alteration of the 5′ cap structure from ARCA to CleanCap was caused by the increasing amount of translatable CAR-mRNA to which the 5′ cap structure was properly added. In addition, mRNAs synthesized using 5-methoxyuridine were reported to be less immunogenic and more efficient in translation [
20]. Although our mRNA coding for CAR#5 did not improve intracellular persistence compared with the coding for CAR#4, CAR#5 showed the highest expression levels early after transfection. These results suggested that the translation rate of CAR#5-coding mRNA is superior to that of CAR#4-coding mRNA containing wild-type uridines. On the other hand, fast translation rates are known to reduce the folding efficiency of nascent proteins [
24]. To be expressed as a functional protein, the nascent protein must be properly folded during biosynthesis in the endoplasmic reticulum and Golgi apparatus; therefore, the protein’s folding efficiency is also regulated by the rate of mRNA translation [
24]. Although the details explaining why the initial expression of CAR#5 could not be maintained are not clearly known, we speculate that the rapid synthesis of artificial CAR#5 proteins may have led to the accumulation of unfolded proteins within the cells, resulting in their elimination (as aberrant proteins).
In addition, a series of experiments on CAR-mRNA construct modification revealed that the removal of the tag sequences in the CAR extracellular domain did not affect the efficiency of CAR expression and activation. Reducing the antigenicity of the CAR extracellular domain to the host immune system is an important consideration, as the appearance of human anti-mouse antibody (HAMA) and human anti-CAR antibody (HACA) can diminish the efficacy of CAR-T cell therapy [
25]. On the contrary, our anti-VEGFR2 scFv (V-85), which was incorporated as the CAR extracellular domain, is a mouse-derived sequence that has not been humanized. Although we believe that scFv should be humanized to completely eliminate the antigenicity of CAR-T cells, the humanization of scFv carries the risk of altering the efficiency of CAR surface expression and antigen recognition properties [
26]. Additionally, clinical trials using non-humanized anti-mesothelin CAR-mRNA transfected CAR-T cells suggested that the appearance of HACA, but not HAMA, limits the efficacy of CAR-T cell therapy. Hence, in the present study, we sought to improve the CAR-mRNA constructs to limit the risk of HACA appearance [
21]. Needless to say, we plan to measure HACA and HAMA against CAR#4-T cells as biomarkers to monitor the efficacy and safety of the TACTICs therapy in the context of the FIH study.
The efficiency of T cell expansion or the functional strength of CAR-T cells is expected to differ depending on the background of individual patients; parameters such as age, complications, and treatment history may influence TACTICs therapy efficacy. Therefore, we have to confirm beforehand if the prepared mRNA-transfected CAR-T cells meet the expected function. Cryopreservation not only allows stockpiling and transporting of functional CAR-T cells, but also offers significant advantages for the assessment of the quality and function of prepared CAR-T cells over time, and immediately before they are administered to patients. In addition, multiple frozen stocks of the same lot allow for the continuous administration of CAR-T cells with consistent quality. However, since the effector function of CAR-T cells was greatly reduced by freeze-thaw procedures, cryopreservation would not be suitable to maintain our mRNA-transfected CAR-T cells. This, together with the notion that cryopreservation reagents available for clinical use are limited, led us to conclude that low-temperature transport (at 4 °C) is the best option for delivering mRNA-transfected CAR-T cells with retained function to patients. On the other hand, we cannot secure a grace period for CAR-T cells quality assessment under 4 °C transportation. Hence, it is necessary to determine the minimum-required parameters, such as viability, CAR expression levels, and CAR integrity, to predict the functionality of CAR-T cells in a short period, and to establish suitability criteria for administration to patients. Furthermore, future analyses using patient-derived T cells would be required to ensure the efficacy of mRNA-transfected anti-VEGFR2 CAR-T cells, since our study was limited to experiments using T cells derived from healthy volunteers.
The present analysis of VEGFR2 expression in frozen STS tissues suggested that TACTICs therapy would be a promising new treatment option for more than 70% of STS patients. Importantly, VEGFR2-positive cases were not characterized by a specific site of origin or STS type, and the levels of vascular invasion and VEGFR2 expression varied widely even within the same STS types. These facts emphasized again the importance of establishing companion diagnostic tools and biomarkers capable of quickly and appropriately selecting patients with VEGFR2-positive STSs, the ones that would benefit the application of TACTICs therapy. We believe that our V-85 antibody would be of great help in predicting the efficacy of TACTICs therapy beforehand. We showed that the V-85 antibody allowed the recognition of VEGFR2; of note, the V-85 antibody prevented the binding anti-VEGFR2 CAR-expressing cells, suggesting both share the same epitope. However, the V-85 antibody is not without limitations: it can only be used in frozen tissue sections, and not in paraffin-embedded ones. Still, and importantly, the commercially available 55B11 antibody showed a VEGFR2 staining potential that correlated with that of the V-85 antibody. Therefore, 55B11 is a useful companion diagnostic reagent applicable to paraffin-embedded biopsy specimens. However, having in mind that the VEGFR2 staining patterns comparing 55B11 and V-85 antibodies differed in some frozen tissue specimens, we cannot exclude possible under/overestimation of VEGFR2 expression in paraffin-embedded tumor and normal specimens. The 55B11 antibody has been widely used in VEGFR2 studies; many results of VEGFR2 expression analysis using this antibody in tumor and normal tissues have been reported [
13,
14,
27]. In The Human Protein Atlas, 55B11 antibody-stained sections are available for different normal tissues, including kidney, colon, testis, skin, esophagus, and bladder tissues; of note, the stained images in tissues other than renal glomeruli were judged to be non-specific responses [
27]. Although the epitope targeted by the 55B11 antibody is unknown, we speculated that 55B11 antibody would react with the intracellular region of some tyrosine kinase molecules, based on the facts that the 55B11 but not the V-85 antibody stained eccrine sweat glands in skin tissues and that various tyrosine kinase inhibitors, including Apatinib (a VEGFR2 tyrosine kinase inhibitor), cause limb syndrome due to eccrine sweat gland injury [
28]. Therefore, VEGFR2 expression analysis using the V-85 antibody in normal and tumor tissues (those stained with the 55B11 antibody) would be required to properly predict the safety of our TACTICs approach and potentially expand its indication to other solid tumors. To achieve this, we may need to follow an approach that combines in vivo administration of the labeled V-85 antibody and imaging diagnosis. Recently, it has been suggested that the therapeutic effect of VEGF/VEGFR2 inhibition in colorectal cancer patients, using an anti-VEGFR2 antibody (Ramucirumab), is linked to the concentration of VEGF-D in peripheral blood [
29]. In the future, tumor environment prediction via liquid biopsies might be possible, reducing the physical burden of invasive biopsy procedures on patients and the risk of cancer cell dissemination. Furthermore, the development of such liquid biopsy-based methods would be advantageous for the determination of both the timing and frequency of mRNA-transfected CAR-T cells administration in our TACTICs therapy. Based on all of the abovementioned, it is essential to estimate the possible sites of adverse events caused by TACTICs therapy and develop countermeasures to safely conduct and execute clinical studies. If we follow this rationale, we believe that a future FIH study using our TACTICs approach will demonstrate both its safety and effectiveness.
4. Materials and Methods
4.1. Cells
Murine L1.2 cells were kindly provided by Prof. Takashi Nakayama (Kindai University, Higashiosaka, Japan) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA). The VEGFR2+ L1.2 cells were generated via retroviral vector transduction (containing the VEGFR2 gene and a puromycin resistance cassette) and were grown in the same culture medium as untransduced L1.2 cells, supplemented with puromycin. NIH/3T3 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). The VEGFR2+ NIH/3T3 cells were also generated via retroviral vector transduction. These NIH/3T3 cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. Human Jurkat cells were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging, and Cancer, Tohoku University (Sendai, Japan). The CAR+ Jurkat cells were also generated via transduction, using a retroviral vector containing the CAR#1 and GFP gene. These Jurkat cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS. All cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
4.2. Construction of CAR-mRNA Variants
The CAR-mRNA constructs used in this study are summarized in
Figure 1A and
Figure S1A. The CAR [V/28/28/28-3z] (CAR#1) and CAR [V/8a/8a/137-3z] constructs [
16] share the same Igκ-chain leader sequence, HA-tag, anti-VEGFR2 scFv (clone V-85) [
30,
31], His-tag, and Flag-tag. CAR#1 has CD28-derived HD/TMD and CD28- and CD3ζ-derived STDs. CAR [V/8a/8a/137-3z] has CD8α-derived HD/TMD and CD137- and CD3ζ-derived STDs. Each construct was subcloned into pcDNA3.1-Zeo (Thermo Fisher Scientific, Waltham, MA, USA). CAR#2 (without the various tag sequences) and CAR#3 (codon-optimized) constructs were generated using artificial gene synthesis (Integrated DNA Technologies, Coralville, IA, USA) and were subcloned into pcDNA3.1-Zeo. Sequence integrity of all plasmids was confirmed using DNA sequencing (Fasmac Co., Atsugi, Japan). Using these plasmids as templates, CAR-mRNAs (#1–#3) containing ARCA as the 5′ cap structure were prepared using the mMESSAGE mMACHINE T7 Ultra kit (Thermo Fisher Scientific, Waltham, MA, USA). CAR#4- or CAR#5-coding mRNAs containing CleanCap as the 5′ cap structure were synthesized using wild-type uridines (CAR#4) or 5-methoxyuridines (CAR#5), considering CAR#3 as the gene template, via Trilink (San Diego, CA, USA).
4.3. Production of CAR-T Cells
Human CAR-T cells were produced as previously described [
16,
32]. Briefly, human T cells derived from healthy volunteers were activated using an anti-CD3 mAb (Janssen Pharmaceutica, Beerse, Belgium) for 13 days, and CAR-mRNAs were introduced into 1 × 10
7 T cells via electroporation using the MaxCyteGT system (MaxCyte, Gaithersburg, MD, USA). Of note, we have confirmed that almost all of the EP-treated T cells were recovered with a high survival rate (cell recovery rate, 90–100%; viability, >97%). The prepared CAR-T cells were maintained in ALyS505N-175 (Cell Science & Technology Institute, Tokyo, Japan) under a humidified atmosphere of 5% CO
2 at 37 °C.
For the study of CAR-T cell freezing, CAR#1-T cells cultured for 3 h after EP treatment were mixed with CP-1 (Kyokuto Pharmaceutical Industrial, Tokyo, Japan) and slowly frozen using a programmed freezer. The frozen CAR-T cells were then rapidly thawed using a water bath set at 37 °C. For the investigation of CAR-T cell preservation under different temperature conditions, CAR#4-T cells cultured for 18 h were stored in a cold storage bag along with a temperature logger at 4 or 18 °C. After 6 h, the cells were collected and subjected to experiments. The viability of each cell line was assessed using the trypan blue exclusion test.
4.4. Reverse Transcription-Quantitative Polymerase Chain Reaction
Total RNA was isolated from CAR-T cells with the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), and reverse-transcribed using the SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). CAR cDNA was detected using Custom TaqMan Gene Expression Assay (Thermo Fisher Scientific, Waltham, MA, USA) targeting the anti-VEGFR2 scFv. CAR and GAPDH expression levels were measured using the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). CAR-mRNA level was normalized to the GAPDH-mRNA level based on the comparative threshold cycle method (2−ΔCt).
4.5. Flow Cytometry Analysis: CAR Surface Expression and VEGFR2-Binding
Human CAR-T cells were incubated with Human TruStain FcX (Biolegend, San Diego, CA, USA) and then stained with a LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific, Waltham, MA, USA) and with PE-Cy7-labeled anti-CD8α mAb (clone HIT8α, Biolegend, San Diego, CA, USA). For CAR expression analysis based on binding to VEGFR2 protein, cells were further stained with recombinant human VEGFR2/KDR Fc chimera protein (VEGFR2-Fc, R&D Systems, Minneapolis, MN, USA) and APC-labeled anti-human IgG Fc mAb (clone HP6017, Biolegend, San Diego, CA, USA). For CAR expression analysis using HA-tag detection, cells were further stained with APC-labeled anti-HA-tag mAb (clone 16B12, Biolegend) or APC-labeled mouse IgG1 isotype control mAb (clone MOPC-21, Biolegend, San Diego, CA, USA). Cells were resolved using BD FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA) or Gallios (Beckman Coulter, Brea, CA, USA) flow cytometers, and data were analyzed using FlowJo software v10 (FlowJo LLC, Ashland, OR, USA). Data were represented as GMFI ratios, calculated according to the following formula: GMFI ratio = (GMFI of CAR-T cells stained with anti-HA mAb or anti-human IgG Fc mAb with VEGFR2-Fc)/(GMFI of CAR-T cells stained with mouse IgG1 isotype control mAb or anti-human IgG Fc mAb without VEGFR2-Fc).
4.6. In Vitro Cytotoxicity Assay
L1.2 cells were labeled with Tag-It Violet Proliferation Cell Tracking Dye (Biolegend, San Diego, CA, USA), and VEGFR2+ L1.2 cells were labeled with Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific, Waltham, MA, USA). mRNA-transfected CAR-T cells 24 h after EP, L1.2 cells, and VEGFR2+ L1.2 cells were co-cultured at the indicated effector cells-to-VEGFR2+ L1.2 cells (E/T) ratios. After 18 h, CountBright Absolute Cell Counting Beads (Thermo Fisher Scientific, Waltham, MA, USA) and 7-AAD Viability Staining Solution (Biolegend, San Diego, CA, USA) were added to the reaction wells, and the number of each target cell was analyzed using flow cytometry until 1000 beads were detected. Cytotoxicity was calculated using the following formula: percentage of VEGFR2-specific lysis = [(VEGFR2+ L1.2 cells/L1.2 cells ratio in the non-effector cells’ well) − (VEGFR2+ L1.2 cells/L1.2 cells ratio in the effector cells’ well)]/[VEGFR2+ L1.2 cells/L1.2 cells ratio in the non-effector cells’ well] × 100.
4.7. In Vivo Cytotoxicity Assay
Female NOG mice (NOD/Shi-scid, IL-2RγΚO Jic) were purchased from In-Vivo Science (Tokyo, Japan) and were maintained in the experimental animal facility at Osaka University. The care and use of laboratory animals complied with the guidelines and policies of the Act on Welfare and Management of Animals in Japan. Protocols and procedures were approved by the Animal Care and Use Committee of Osaka University.
A mixture of 2 × 106 VEGFR2+ L1.2 cells pre-stained with the Cell Proliferation Dye eFluor 670 and 1 × 106 L1.2 cells pre-stained with the Tag-It Violet Proliferation Cell Tracking Dye were injected into the orbital venous plexus of NOG mice. Two days later, 1 × 107 CAR#4-T cells or Mock-T cells (6 h after 4 °C preservation) were administered to NOG mice via the same route. Two days later, the NOG mice were euthanized and VEGFR2+ L1.2 cells and L1.2 cells in their spleens were detected using a flow cytometer. In vivo killing activity of CAR-T cells against VEGFR2+ L1.2 cells was calculated using the following formula: percentage of VEGFR2-specific in vivo cytotoxicity = [(VEGFR2+ L1.2 cells/L1.2 cells ratio in the vehicle group) − (VEGFR2+ L1.2 cells/L1.2 cells ratio in the experimental group)]/[VEGFR2+ L1.2 cells/L1.2 cells ratio in the vehicle group] × 100.
4.8. Generation of an Antibody Recognizing the Same VEGFR2 Extracellular Domain as CAR
A rabbit antibody (V-85 antibody) with the same variable region as that of anti-VEGFR2 scFv in CAR was obtained from Syd Labs (Boston, MA, USA). To evaluate the VEGFR2 binding properties of this antibody, we performed immunofluorescence and flow cytometry analysis using VEGFR2+ NIH/3T3 cells.
For immunofluorescence, NIH/3T3 cells and VEGFR2+ NIH/3T3 cells cultured on glass slides were fixed in PBS containing 4% paraformaldehyde and then stained with the V-85 antibody and Alexa Fluor 647-goat anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody (Thermo Fisher Scientific), or anti-human CD309 (VEGFR2) antibody (clone 7D4-6, Biolegend, San Diego, CA, USA) and Alexa Fluor 647-goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody (Thermo Fisher Scientific). Afterward, the antibody solution was thoroughly removed, and the preparations were sealed with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA). Fluorescence images were acquired using a BZ-X800 fluorescence microscope (Keyence Corporation, Osaka, Japan). For flow cytometric analysis, cells were suspended, stained with the same set of antibodies, and analyzed using a BD FACS Canto II flow cytometer.
4.9. Cell-Binding Assay Using VEGFR2+ NIH/3T3 and CAR+ Jurkat Cells
Jurkat or CAR+ Jurkat cells were labeled with Cell Proliferation Dye eFluor 670. 1 × 106 Jurkat cells (per condition) were then added to NIH/3T3 or VEGFR2+ NIH/3T3 cells fixed in sheets on glass slides and incubated at 37 °C for 6 h. The Jurkat cell suspensions were thoroughly removed and then the preparations were sealed with ProLong Diamond Antifade Mountant with DAPI. Fluorescence images were acquired using a BZ-X800 fluorescence microscope, and then, the Jurkat cells on the slide were counted for four fields of view. In the competitive inhibition assay using these cell lines (VEGFR2+ NIH/3T3 cells and CAR+ Jurkat cells), NIH/3T3 cells were first incubated with two anti-VEGFR2 antibodies (V-85 or 7D4-6; 25 ng/mL) overnight at 4 °C, and only then co-cultured with Jurkat cells.
4.10. Immunofluorescence Staining of Frozen Tissue Specimens
This study was conducted after receiving approval from the Observational Research Ethics Review Committee of the Osaka University Hospital. STS and surrounding normal tissue (muscle, adipose or skin) blocks were obtained after extensive resections from STS patients and were fixed via immersion in PBS containing 4% paraformaldehyde for 24 h. Preparation of frozen tissue blocks, sectioning and hematoxylin and eosin (H&E) staining were performed by the Applied Medical Research Laboratory (Osaka, Japan).
Frozen tissue specimens were blocked with PBS containing 5% goat serum and 0.3% Triton X-100 at room temperature for 1 h. The blocking buffer was removed, and the anti-VEGFR2 mAb (V-85 antibody or 55B11 antibody (Cell Signaling Technology, Danvers, MA, USA)) as well as the purified anti-human CD31 antibody (clone WM59, BioLegend, San Diego, CA, USA) in antibody dilution buffer (PBS containing 1% BSA and 0.3% Triton X-100) were added and allowed to react overnight at 4 °C. After removal of the antibody solution, preparations were thoroughly washed with TBS containing 0.05% Tween-20. Then, the Alexa Fluor 488-goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) and the Alexa Fluor 647-goat anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody mixture was prepared in antibody dilution buffer, added to the preparations, and allowed to react for 2 h at room temperature. Again, the antibody solution was thoroughly removed, and the preparations were sealed with ProLong Diamond Antifade Mountant with DAPI. Fluorescence images were acquired using a BZ-X800 fluorescence microscope and the brightness (MFI) of each protein detection was measured using the BZ-H4M/Measurement application. Data were represented as ΔMFI of tissues stained with primary and corresponding secondary antibodies to each antigen, minus MFI of tissues stained with secondary antibodies only.
4.11. Immunohistochemical Staining of Paraffin-Embedded Specimens
This study was conducted after receiving approval from the Observational Research Ethics Review Committee of the Osaka University Hospital and Hokkaido University Hospital. Tissue microarray blocks were constructed using a manual tissue microarrayer (JF-4; Sakura Finetek Japan, Tokyo, Japan) with a 2.0-mm diameter needle, from two representative tumor areas (both the invasive front and the bulk of the tumor) and from one representative area of non-neoplastic bile duct as an internal control. The finalized array blocks were sliced into 4-μm-thick sections and mounted onto glass slides. Tissue sections were deparaffinized in xylene and rehydrated through a series of graded ethanol solutions. Heat-induced antigen retrieval was carried out in high-pH antigen retrieval buffer (Dako Cytomation, Glostrup, Denmark). Endogenous peroxidase was quenched with 3% H2O2 for 5 min. These sections were visualized using the HRP-labeled polymer method (EnVision FLEX system, Dako Cytomation). Immunostained sections were counterstained with hematoxylin, dehydrated in ethanol, and cleared in xylene.
4.12. Statistical Analysis
The results of the CAR expression profile and cytotoxicity analysis and cell-binding assay are represented as the mean ± SD. Statistical analysis of these data was performed using Student’s t-test for a two-group comparison and Dunnet’s test or Tukey’s test for multi-group comparisons using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA). The results of in vivo cytotoxic assay are represented ± SE, and their statistical analysis was performed using Welch’s t-test. A p value lower that 0.05 was considered statistically significant.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




