The Communication between the PI3K/AKT/mTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer

 ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Clinical Investigation into mTOR Signaling and Inhibition in Women with Gynecological Cancers
2.1. Endometrial Cancer
2.2. Cervical Cancer
2.3. Ovarian Cancer
2.4. Ongoing Clinical Trials
3. The PI3K/AKT/mTOR Impact on Molecular Pathophysiology of Gynecological Cancer
3.1. PI3K/AKT/mTOR Pathway
3.2. mTORC1 and mTORC2 Complexes in Gynecological Malignancies
3.3. Metabolic Impact on Carcinogenesis by mTOR
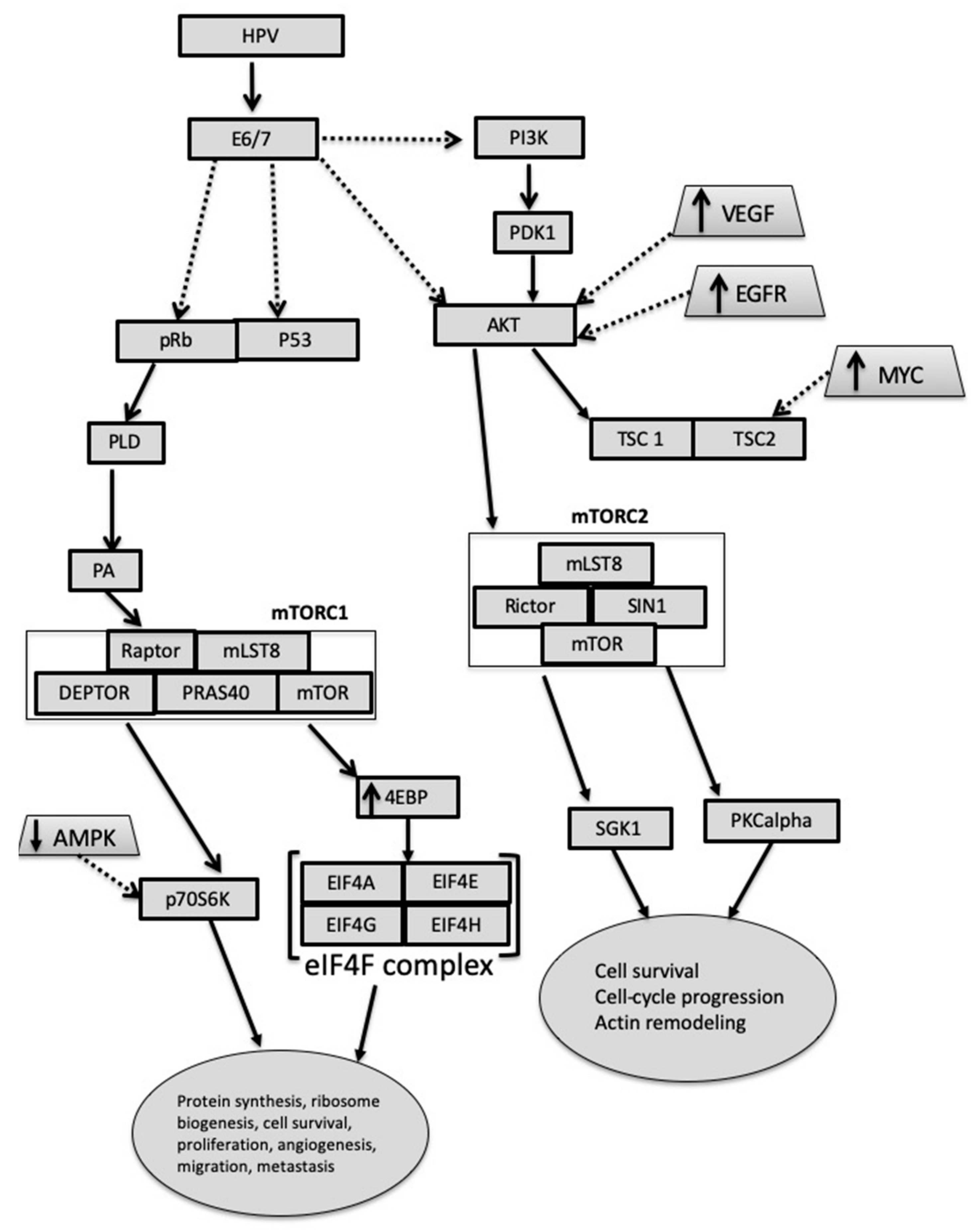
3.4. Carcinogenesis in Presence and Absence of HPV
3.5. Growth Factors in mTOR Dysregulation
3.6. Genomic Aberrations Interacting with mTOR Signaling Pathway Activation
3.7. Epithelial-Mesenchymal Transition and mTOR Signaling
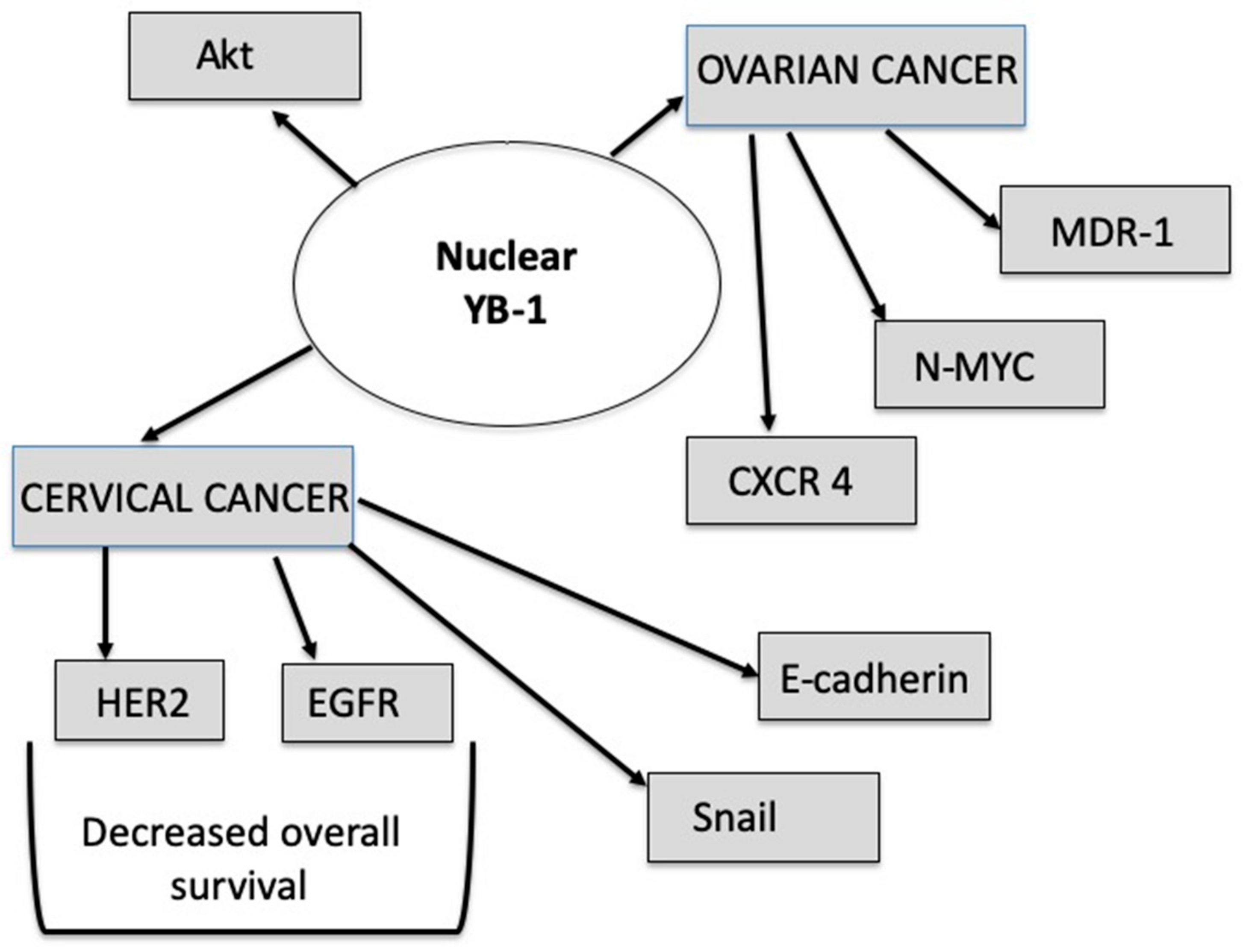
3.8. RNA-Based Alterations Interacting with mTOR
3.9. Hypoxic Modulation of Carcinogenesis via mTOR Signaling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Husseinzadeh, N.; Husseinzadeh, H.D. MTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: A critical review. Gynecol. Oncol. 2014, 133, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.; Landen, C. The importance of the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 8213–8227. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Kurokawa, T.; Horiuchi, Y.; Sawamura, Y.; Shinagawa, A.; Kotsuji, F. Localisation of phosphorylated mTOR expression is critical to tumour progression and outcomes in patients with endometrial cancer. Eur. J. Cancer 2010, 46, 3445–3452. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, J.A.; Mertens, P.R. Cold shock proteins: From cellular mechanisms to pathophysiology and disease. Cell Commun. Signal. 2018, 16, 1–14. [Google Scholar] [CrossRef]
- Nishio, S.; Ushijima, K.; Yamaguchi, T.; Sasajima, Y.; Tsuda, H.; Kasamatsu, T.; Kage, M.; Ono, M.; Kuwano, M.; Kamura, T. Nuclear Y-box-binding protein-1 is a poor prognostic marker and related to epidermal growth factor receptor in uterine cervical cancer. Gynecol. Oncol. 2014, 132, 703–708. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Brandt, S.; Bernhardt, A.; Gorny, X.; Schindele, D.; Jandrig, B.; Schostak, M.; Isermann, B.; Lindquist, J.A.; Mertens, P.R. Crosstalk between Akt signaling and cold shock proteins in mediating invasive cell phenotypes. Oncotarget 2018, 9, 19039–19049. [Google Scholar] [CrossRef]
- Maier, E.; Attenberger, F.; Tiwari, A.; Lettau, K.; Rebholz, S.; Fehrenbacher, B.; Schaller, M.; Gani, C.; Toulany, M. Dual targeting of Y-box binding Protein-1 and Akt inhibits proliferation and enhances the chemosensitivity of colorectal cancer cells. Cancers 2019, 11, 562. [Google Scholar] [CrossRef]
- Darb-Esfahani, S.; Faggad, A.; Noske, A.; Weichert, W.; Buckendahl, A.C.; Müller, B.; Budczies, J.; Röske, A.; Dietel, M.; Denkert, C. Phospho-mTOR and phospho-4EBP1 in endometrial adenocarcinoma: Association with stage and grade in vivo and link with response to rapamycin treatment in vitro. J. Cancer Res. Clin. Oncol. 2009, 135, 933–941. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Duran, I.; Clarke, B.A.; Oza, A.M. Biologic rationale and clinical activity of mTOR inhibitors in gynecological cancer. Cancer Treat. Rev. 2012, 38, 767–775. [Google Scholar] [CrossRef]
- Kato, E.; Orisaka, M.; Kurokawa, T.; Chino, Y.; Fujita, Y.; Shinagawa, A.; Yoshida, Y. Relation between outcomes and expression of estrogen receptor-α phosphorylated at Ser167 in endometrioid endometrial cancer. Cancer Sci. 2014, 105, 1307–1312. [Google Scholar] [CrossRef]
- Kassem, L.; Abdel-Rahman, O. Targeting mTOR pathway in gynecological malignancies: Biological rationale and systematic review of published data. Crit. Rev. Oncol. Hematol. 2016, 108, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.G.T.; Krampe, J.; Ruhland, B.; Diedrich, K.; Hornung, D.; Agic, A. Cold-shock domain family member YB-1 expression in endometrium and endometriosis. Hum. Reprod. 2012, 27, 173–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, R.; Yang, Z.; Xu, G.; Yu, S. miR-338 modulates proliferation and autophagy by PI3K/AKT/mTOR signaling pathway in cervical cancer. Biomed. Pharmacother. 2018, 105, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, H.Y.; Zhang, P.N.; Lu, X.; Sun, H. Elevated phosphatidylinositol 3-kinase activation and its clinicopathological significance in cervical cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 139, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zheng, P.S. Activation of mTOR signaling pathway contributes to survival of cervical cancer cells. Gynecol. Oncol. 2010, 117, 103–108. [Google Scholar] [CrossRef]
- Feng, W.; Duan, X.; Liu, J.; Xiao, J.; Brown, R.E. Morphoproteomic evidence of constitutively activated and overexpressed mTOR pathway in cervical squamous carcinoma and high grade squamous intraepithelial lesions. Int. J. Clin. Exp. Pathol. 2009, 2, 249–260. [Google Scholar]
- De Melo, A.C.; Paulino, E.; Garces, A.H.I. A review of mTOR pathway inhibitors in gynecologic cancer. Oxid. Med. Cell. Longev. 2017, 2017, 4809751. [Google Scholar] [CrossRef]
- Zhang, Y.; Reng, S.R.; Wang, L.; Lu, L.; Zhao, Z.H.; Zhang, Z.K.; Feng, X.D.; Ding, X.D.; Wang, J.; Feng, G.; et al. Overexpression of Y-box binding protein-1 in cervical cancer and its association with the pathological response rate to chemoradiotherapy. Med. Oncol. 2012, 29, 1992–1997. [Google Scholar] [CrossRef]
- Pang, T.; Li, M.; Zhang, Y.; Yong, W.; Kang, H.; Yao, Y.; Hu, X. Y Box-binding protein 1 promotes epithelial-mesenchymal transition, invasion, and metastasis of cervical cancer via enhancing the expressions of snail. Int. J. Gynecol. Cancer 2017, 27, 1753–1760. [Google Scholar] [CrossRef]
- Smolle, E.; Taucher, V.; Pichler, M.; Petru, E.; Lax, S.; Haybaeck, J. Targeting signaling pathways in epithelial ovarian cancer. Int. J. Mol. Sci. 2013, 14, 9536–9555. [Google Scholar] [CrossRef]
- MacKay, H.J.; Eisenhauer, E.A.; Kamel-Reid, S.; Tsao, M.; Clarke, B.; Karakasis, K.; Werner, H.M.J.; Trovik, J.; Akslen, L.A.; Salvesen, H.B.; et al. Molecular determinants of outcome with mammalian target of rapamycin inhibition in endometrial cancer. Cancer 2014, 120, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Rohr, I.; Braicu, E.I.; En-Nia, A.; Heinrich, M.; Richter, R.; Chekerov, R.; Dechend, R.; Heidecke, H.; Dragun, D.; Schäfer, R.; et al. Y-box protein-1/p18 as novel serum marker for ovarian cancer diagnosis: A study by the Tumor Bank Ovarian Cancer (TOC). Cytokine 2016, 85, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Marneth, A.E.; Alexe, G.; Walker, S.R.; Gandler, H.I.; Ye, D.Q.; Labella, K.; Mathur, R.; Toniolo, P.A.; Tillgren, M.; et al. The kinases IKBKE and TBK1 regulate MYC-dependent survival pathways through YB-1 in AML and are targets for therapy. Blood Adv. 2018, 2, 3428–3442. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jiao, X.; Jiao, J.; Zhang, T.; Cui, B. Increased expression of FHL2 promotes tumorigenesis in cervical cancer and is correlated with poor prognosis. Gene 2018, 669, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, P.; Wei, W. mTOR signaling in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Montero, J.C.; Chen, X.; Ocaña, A.; Pandiella, A. Predominance of mTORC1 over mTORC2 in the regulation of proliferation of ovarian cancer cells: Therapeutic implications. Mol. Cancer Ther. 2012, 11, 1342–1352. [Google Scholar] [CrossRef]
- Noske, A.; Lindenberg, J.L.; Darb-Esfahani, S.; Weichert, W.; Buckendahl, A.-C.; Röske, A.; Sehouli, J.; Dietel, M.; Denkert, C. Activation of mTOR in a subgroup of ovarian carcinomas: Correlation with p-eIF-4E and prognosis. Oncol. Rep. 2008, 20, 1409–1417. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Lyabin, D.N.; Ovchinnikov, L.P. Selective regulation of YB-1 mRNA translation by the mTOR signaling pathway is not mediated by 4E-binding protein. Sci. Rep. 2016, 6, 22502. [Google Scholar] [CrossRef]
- Iglesias, D.A.; Zhang, Q.; Celestino, J.; Sun, C.C.; Yates, M.S.; Schmandt, R.E.; Lu, K.H. Lean body weight and metformin are insufficient to prevent endometrial hyperplasia in mice harboring inactivating mutations in PTEN. Oncology 2017, 92, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.S.; Lombard, J.M.; Ius, Y.; O’Sullivan, R.; Wood, L.G.; Nahar, P.; Jaaback, K.; Tanwar, P.S. Adipose-derived VEGF–mTOR signaling promotes endometrial hyperplasia and cancer: Implications for obese women. Mol. Cancer Res. 2018, 16, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shen, J.; Gao, L.; Feng, Y. Estrogen promotes fat mass and obesity-associated protein nuclear localization and enhances endometrial cancer cell proliferation via the mTOR signaling pathway. Oncol. Rep. 2016, 35, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, L.; Guo, H.; Wysham, W.Z.; Roque, D.R.; Willson, A.K.; Sheng, X.; Zhou, C.; Bae-Jump, V.L. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol. Oncol. 2015, 138, 668–675. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer. 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Inagawa, Y.; Yoshimatsu, Y.; Haga, K.; Tanaka, K.; Egawa, N.; Ohno, S.I.; Ichikawa, H.; Yugawa, T.; Fujita, M.; et al. A critical role of MYC for transformation of human cells by HPV16 E6E7 and oncogenic HRAS. Carcinogenesis 2012, 33, 910–917. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Marsh, C.; El Dinali, M.; Gangane, N.; Jennison, K.; Hewitt, S.; Patel, V.; Seiwert, T.Y.; Gutkind, J.S. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clin. Cancer Res. 2012, 18, 2558–2568. [Google Scholar] [CrossRef]
- Rabachini, T.; Boccardo, E.; Andrade, R.; Perez, K.R.; Nonogaki, S.; Cuccovia, I.M.; Villa, L.L. HPV-16 E7 expression up-regulates phospholipase D activity and promotes rapamycin resistance in a pRB-dependent manner. BMC Cancer 2018, 18, 3–10. [Google Scholar] [CrossRef]
- Ueno, S.; Sudo, T.; Oka, N.; Wakahashi, S.; Yamaguchi, S.; Fujiwara, K.; Mikami, Y.; Nishimura, R. Absence of human papillomavirus infection and activation of pi3k-akt pathway in cervical clear cell carcinoma. Int. J. Gynecol. Cancer 2013, 23, 1084–1091. [Google Scholar] [CrossRef]
- Roche, E.; Lascombe, I.; Bittard, H.; Mougin, C.; Fauconnet, S. The PPARβ agonist L-165041 promotes VEGF mRNA stabilization in HPV18-harboring HeLa cells through a receptor-independent mechanism. Cell. Signal. 2014, 26, 433–443. [Google Scholar] [CrossRef]
- Grassi, M.L.; de Souza Palma, C.; Thomé, C.H.; Lanfredi, G.P.; Poersch, A.; Faça, V.M. Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control. J. Proteom. 2017, 151, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Trinh, X.B.; Tjalma, W.A.A.; Vermeulen, P.B.; Van den Eynden, G.; Van der Auwera, I.; Van Laere, S.J.; Helleman, J.; Berns, E.M.J.J.; Dirix, L.Y.; van Dam, P.A. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br. J. Cancer 2009, 100, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.T.; Leung, P.C. The PI3K/Akt/mTOR signaling pathway mediates insulin-like growth factor 1-induced E-cadherin down-regulation and cell proliferation in ovarian cancer cells. Cancer Lett. 2012, 326, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Griner, S.E.; Joshi, J.P.; Nahta, R. Growth differentiation factor 15 stimulates rapamycin-sensitive ovarian cancer cell growth and invasion. Biochem. Pharmacol. 2013, 85, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.B.; Byun, H.J.; Park, S.H.; Park, C.Y.; Lee, S.H.; Rho, S.B. CYR61 controls p53 and NF-κB expression through PI3K/Akt/mTOR pathways in carboplatin-induced ovarian cancer cells. Cancer Lett. 2012, 315, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, C.; Dong, P.; Guo, Y.; Mu, N. Molecular regulation of cervical cancer growth and invasion by VEGFa. Tumor Biol. 2014, 35, 11587–11593. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef]
- Aust, S.; Auer, K.; Bachmayr-Heyda, A.; Denkert, C.; Sehouli, J.; Braicu, I.; Mahner, S.; Lambrechts, S.; Vergote, I.; Grimm, C.; et al. Ambivalent role of pFAK-Y397 in serous ovarian cancer-a study of the OVCAD consortium. Mol. Cancer 2014, 13, 1–11. [Google Scholar] [CrossRef]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Ogishima, J.; et al. Targeting glutamine metabolism and the focal adhesion kinase additively inhibits the mammalian target of the rapamycin pathway in spheroid cancer stem-like properties of ovarian clear cell carcinoma in Vitro. Int. J. Oncol. 2017, 50, 1431–1438. [Google Scholar] [CrossRef]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Adenan, N.A.M.; Chung, I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 2013, 8, 1–16. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Squillace, R.M.; Wongchenko, M.J.; Miller, D.; Wardwell, S.; Mohemmad, Q.; Narasimhan, N.I.; Wang, F.; Clackson, T.; Rivera, V.M. Combined targeting of FGFR2 and mTOR by ponatinib and ridaforolimus results in synergistic antitumor activity in FGFR2 mutant endometrial cancer models. Cancer Chemother Pharmacol. 2013, 71, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; De Vivo, I.; Huang, S.; Zhu, X.; Risch, H.; Moore, J.H.; Yu, H.; Garmire, L.X. Meta-dimensional data integration identifies critical pathways for susceptibility, tumorigenesis and progression of endometrial cancer. Oncotarget 2016, 7, 55249. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Painter, J.N.; Kaufmann, S.; O’Mara, T.A.; Hillman, K.M.; Sivakumaran, H.; Darabi, H.; Cheng, T.H.T.; Pearson, J.; Kazakoff, S.; Waddell, N.; et al. A Common Variant at the 14q32 Endometrial Cancer Risk Locus Activates AKT1 through YY1 Binding. Am. J. Hum. Genet. 2016, 98, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Huang, H.s.D.; Yeh, K.T.; Chang, J.G. Genetic alterations in endometrial cancer by targeted next-generation sequencing. Exp. Mol. Pathol. 2016, 100, 8–12. [Google Scholar] [CrossRef]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; De Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar] [CrossRef]
- Wang, Y.; Sheng, Q.; Spillman, M.A.; Behbakht, K.; Gu, H. Gab2 regulates the migratory behaviors and E-cadherin expression via activation of the PI3K pathway in ovarian cancer cells. Oncogene 2012, 31, 2512–2520. [Google Scholar] [CrossRef]
- Duckworth, C.; Zhang, L.; Carroll, S.L.; Ethier, S.P.; Cheung, H.W. Overexpression of GAB2 in ovarian cancer cells promotes tumor growth and angiogenesis by upregulating chemokine expression. Oncogene 2016, 35, 4036–4047. [Google Scholar] [CrossRef]
- Cheng, K.; Hao, M. Mammalian Target of Rapamycin (mTOR) Regulates Transforming Growth Factor-β1 (TGF-β1)-Induced Epithelial-Mesenchymal Transition via Decreased Pyruvate Kinase M2 (PKM2) Expression in Cervical Cancer Cells. Med. Sci. Monit. 2017, 23, 2017–2028. [Google Scholar] [CrossRef]
- Li, Y.; Wu, T.; Wang, Y.; Yang, L.; Hu, C.; Chen, L.; Wu, S. γ-Glutamyl cyclotransferase contributes to tumor progression in high grade serous ovarian cancer by regulating epithelial-mesenchymal transition via activating PI3K/AKT/mTOR pathway. Gynecol. Oncol. 2018, 149, 163–172. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Sun, H. Up-regulation of Foxp3 inhibits cell proliferation, migration and invasion in epithelial ovarian cancer. Cancer Lett. 2010, 287, 91–97. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, B.; Wang, H.Y.; Chang, A.; Zheng, X.F.S. Emerging role of MicroRNAs in mTOR signaling. Cell. Mol. Life Sci. 2017, 74, 2613–2625. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Ahmad, A.; Zubair, H.; Miree, O.; Singh, S.; Rocconi, R.P.; Scalici, J.; Singh, A.P. MicroRNAs in gynecological cancers: Small molecules with big implications. Cancer Lett. 2017, 407, 123–138. [Google Scholar] [CrossRef]
- Nair, V.B.; Manasa, V.G.; Sinto, M.S.; Jayasree, K.; James Francis, V.; Kannan, S. Differential expression of MicroRNAs in uterine cervical cancer and its implications in carcinogenesis; an integrative approach. Int. J. Gynecol. Cancer 2018, 28, 553–562. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, K.X.; Xu, H.; Hong, Y. Integrative miRNA analysis identifies hsa-miR-3154, hsa-miR-7-3, and hsa-miR-600 as potential prognostic biomarker for cervical cancer. J. Cell. Biochem. 2018, 119, 1558–1566. [Google Scholar] [CrossRef]
- Torres, A.; Torres, K.; Pesci, A.; Ceccaroni, M.; Paszkowski, T.; Cassandrini, P.; Zamboni, G.; Maciejewski, R. Deregulation of miR-100, miR-99a and miR-199b in tissues and plasma coexists with increased expression of mTOR kinase in endometrioid endometrial carcinoma. BMC Cancer 2012, 12, 369. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, M.; Li, Q.; Zhu, P. MiR-101 reduces cell proliferation and invasion and enhances apoptosis in endometrial cancer via regulating PI3K/Akt/mTOR. Cancer Biomark. 2017, 21, 189–196. [Google Scholar] [CrossRef]
- Zhuo, Z.; Yu, H. miR-205 inhibits cell growth by targeting AKT-mTOR signaling in progesterone-resistant endometrial cancer Ishikawa cells. Oncotarget 2017, 8, 28042–28051. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Zhang, X.; Lin, Y.; Luo, T.; Xiao, Z.; Zhou, Q. A dual PI3K/AKT/mTOR signaling inhibitor miR-99a suppresses endometrial carcinoma. Am. J. Transl. Res. 2016, 8, 719–731. [Google Scholar]
- Wu, D.; Huang, H.-J.; He, C.-N.; Wang, K.-Y. MicroRNA-199a-3p regulates endometrial cancer cell proliferation by targeting mammalian target of rapamycin (mTOR). Int. J. Gynecol. Cancer 2013, 23, 1191–1197. [Google Scholar] [CrossRef]
- Tsuruta, T.; Kozaki, K.I.; Uesugi, A.; Furuta, M.; Hirasawa, A.; Imoto, I.; Susumu, N.; Aoki, D.; Inazawa, J. miR-152 is a tumor suppressor microRNA that is silenced by DNA hypermethylation in endometrial cancer. Cancer Res. 2011, 71, 6450–6462. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, W.; Huang, N.; Yang, B.B.; Cui, K.; Wan, G.; Liao, M.; He, J.; Xu, H.; Lao, Y.; et al. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy 2013, 10, 70–79. [Google Scholar] [CrossRef]
- Wang, F.; Shan, S.; Huo, Y.; Xie, Z.; Fang, Y.; Qi, Z.; Chen, F.; Li, Y.; Sun, B. MiR-155-5p inhibits PDK1 and promotes autophagy via the mTOR pathway in cervical cancer. Int. J. Biochem. Cell Biol. 2018, 99, 91–99. [Google Scholar] [CrossRef]
- Wang, L.; Chang, L.; Li, Z.; Gao, Q.; Cai, D.; Tian, Y.; Zeng, L.; Li, M. MiR-99a and -99b inhibit cervical cancer cell proliferation and invasion by targeting mTOR signaling pathway. Med. Oncol. 2014, 31, 934. [Google Scholar] [CrossRef]
- Cong, J.; Liu, R.; Wang, X.; Jiang, H.; Zhang, Y. MiR-634 decreases cell proliferation and induces apoptosis by targeting mTOR signaling pathway in cervical cancer cells. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1694–1701. [Google Scholar] [CrossRef]
- Dai, C.; Xie, Y.; Zhuang, X.; Yuan, Z. MiR-206 inhibits epithelial ovarian cancer cells growth and invasion via blocking c-Met/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2018, 104, 763–770. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Tang, W.; Lin, Z.; Xu, L.; Dong, R.; Li, Y.; Li, J.; Zhang, Z.; Li, X.; et al. MiR-130a upregulates mTOR pathway by targeting TSC1 and is transactivated by NF-? B in high-grade serous ovarian carcinoma. Cell Death Differ. 2017, 24, 2089–2100. [Google Scholar] [CrossRef]
- Wang, Z.; Ting, Z.; Li, Y.; Chen, G.; Lu, Y.; Hao, X. microRNA-199a is able to reverse cisplatin resistance in human ovarian cancer cells through the inhibition of mammalian target of rapamycin. Oncol. Lett. 2013, 6, 789–794. [Google Scholar] [CrossRef]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A Link between mir-100 and FRAP1/mTOR in Clear Cell Ovarian Cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef]
- Wang, J.Y.; Lu, A.Q.; Chen, L.J. LncRNAs in ovarian cancer. Clin Chim. Acta 2019, 490, 17–27. [Google Scholar] [CrossRef]
- Xie, P.; Cao, H.; Li, Y.; Wang, J.; Cui, Z. Knockdown of lncRNA CCAT2 inhibits endometrial cancer cells growth and metastasis via sponging miR-216b. Cancer Biomark. 2017, 21, 123–133. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Wang, J.; Wang, Y.; Liu, L.; Xu, X. LncRNA MEG3 has anti-activity effects of cervical cancer. Biomed. Pharmacother. 2017, 94, 636–643. [Google Scholar] [CrossRef]
- Xuan, K.; Dan, S.; Wu, D.; Chen, S.; Zhao, Y.; Hong, Z. LncRNA MEG3 inhibit endometrial carcinoma tumorigenesis and progression through PI3K pathway. Apoptosis 2017, 22, 1543–1552. [Google Scholar]
- Xiu, Y.L.; Sun, K.X.; Chen, X.; Chen, S.; Zhao, Y.; Guo, Q.G.; Zong, Z.H. Upregulation of the lncRNA Meg3 induces autophagy to inhibit tumorigenesis and progression of epithelial ovarian carcinoma by regulating activity of ATG3. Oncotarget 2017, 8, 31714–31725. [Google Scholar] [CrossRef]
- Du, Y.; Wang, L.; Chen, S.; Liu, Y.; Zhao, Y. lncRNA DLEU1 contributes to tumorigenesis and development of endometrial carcinoma by targeting mTOR. Mol. Carcinog. 2018, 57, 1191–1200. [Google Scholar] [CrossRef]
- Liu, C.; Tian, X.; Zhang, J.; Jiang, L. Long Non-coding RNA DLEU1 promotes proliferation and invasion by interacting with miR-381 and enhancing HOXA13 expression in cervical cancer. Front. Genet. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Wang, L.L.; Sun, K.X.; Wu, D.D.; Xiu, Y.L.; Chen, X.; Chen, S.; Zong, Z.H.; Sang, X.B.; Liu, Y.; Zhao, Y. DLEU1 contributes to ovarian carcinoma tumourigenesis and development by interacting with miR-490-3p and altering CDK1 expression. J. Cell. Mol. Med. 2017, 21, 3055–3065. [Google Scholar] [CrossRef]
- Zhang, S.; Leng, T.; Zhang, Q.; Zhao, Q.; Nie, X.; Yang, L. Sanguinarine inhibits epithelial ovarian cancer development via regulating long non-coding RNA CASC2-EIF4A3 axis and/or inhibiting NF-κB signaling or PI3K/AKT/mTOR pathway. Biomed. Pharmacother. 2018, 102, 302–308. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Sahasrabudhe, N.M.; McGregor, F.; Chalmers, A.J.; Cassidy, J.; Plumb, J. Hypoxia-inducible factor 1 alpha is required for the tumourigenic and aggressive phenotype associated with Rab25 expression in ovarian cancer. Oncotarget 2016, 7, 22650–22664. [Google Scholar] [CrossRef]
- Kim, H.; Woo, D.J.; Kim, S.Y.; Yang, E.G. p21-activated kinase 4 regulates HIF-1α translation in cancer cells. Biochem. Biophys. Res. Commun. 2017, 486, 270–276. [Google Scholar] [CrossRef]
- Ivanova, I.G.; Park, C.V.; Yemm, A.I.; Kenneth, N.S. PERK/eIF2α signaling inhibits HIF-induced gene expression during the unfolded protein response via YB1-dependent regulation of HIF1α translation. Nucleic Acids Res. 2018, 46, 3878–3890. [Google Scholar] [CrossRef]
- Kim, H.; Na, Y.R.; Kim, S.Y.; Yang, E.G. Protein kinase C isoforms differentially regulate hypoxia-inducible factor-1α accumulation in cancer cells. J. Cell. Biochem. 2016, 117, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.N.; Yang, H.; Huang, G.Y.; Fu, C.; Pontikos, M.; Wang, Y.; Mao, W.; Pang, L.; Yang, M.; Liu, J.; et al. RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 2018, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumour dormancy in human ovarian cancer cells. Cell Prolif. 2008, 118, 3917–3929. [Google Scholar]
- Bossler, F.; Kuhn, B.J.; Günther, T.; Kraemer, S.J.; Khalkar, P.; Adrian, S.; Lohrey, C.; Holzer, A.; Shimobayashi, M.; Dürst, M.; et al. Repression of human papillomavirus oncogene expression under hypoxia is mediated by PI3K/mTORC2/AKT signaling. MBio 2019, 10, e02323-18. [Google Scholar] [CrossRef] [PubMed]
- Bossler, F.; Hoppe-Seyler, K.; Hoppe-Seyler, F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2188. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Therapeutic Agent | Carcinoma | Trial Drug Design | Reg. Nr. |
|---|---|---|---|---|
| mTORC1 | everolimus | EC | Everolimus (RAD001) and Letrozole or Hormonal Therapy | NCT02228681 |
| Atypical hyperplasia or FIGO stage IA EC | Levonorgestrel-Releasing Intrauterine System with or without Everolimus | NCT02397083 | ||
| OC, EC, CC | Patients with alterations in PIK3CA, PIK3R1, AKT1, AKT2, mTOR, RICTOR, RAPTOR genes, or with TSC1, TSC2 or PTEN loss for maintenance therapy | NCT02029001 | ||
| everolimus temsirolimus | Advanced OC | Vemurafenib in Combination with Everolimus or Temsirolimus | NCT01596140 | |
| sirolimus | Stage II-IV OC | The effects of TRICOM vaccine with sirolimus | NCT01536054 | |
| mTORC1/2 | Vistusertib (AZD2014) | HR positive EC | AZD2014 and anastrozole vs. anastrozole alone | NCT02730923 |
| Sapanisertib MLN0128 | EC and other solid tumors | Single experimental arm: bevacizumab and MLN0128 | NCT02142803 | |
| Sapanisertib MLN0128 | EC, OC | Patients with mTOR mutation receive sapanisertib as a single experimental arm | NCT02465060 | |
| AKT inhibitors | miransertib ARQ 092 | OC, CC, EC | ARQ 092 + paclitaxel or ARQ 092 + anastrozole | NCT02476955 |
| PI3K inhibition | Copanlisib Hydrochloride | CC, OC, EC | Patients with a PI3K or a PTEN mutation receive copanlisib | NCT02465060 |
| GSK2636771 | CC, OC, EC | Patients with a PTEN mutation/expression/loss | NCT02465060 | |
| Dual PI3K/mTOR inhibition | Gedatolisib PF-05212384 | EC and other solid tumors | Single experimental arm: palbociclib and gedatolisib | NCT03065062 |
| mTORC1/2 vs. AKT inhibition | vistusertib AZD2014 vs. capivasertib AZD5363 | Recurrent EC or OC | (olaparib, vistusertib) vs. (olaparib, capivasertib) | NCT02208375 |
| Dual Akt/ERK inhibition | ONC201 | Recurrent or metastatic EC | Single experimental arm: ONC201 treatment | NCT03099499 |
| Type of Investigated Cancer | miRNA | Component | Impact of Aberration on Signaling |
|---|---|---|---|
| Endometrial cancer | miR-101 [66] | mTORC1/mTORC2 | Downregulation leads to mTOR upregulation |
| miR-205 [67] | PTEN expression regulation | Low levels of miR-205 lead to reduced pmTOR and pAKT expression | |
| miR99 family [65,68] | PI3K through direct target IGF/IGFR | Low levels of expression correlated with better tumor differentiation | |
| miR-199a-3p [69] | mTORC1/mTORC2 | Upregulation inhibits tumor cell proliferation through negative regulation of mTOR expression | |
| miR-199b [65] | mTOR kinase | High expression in better differentiated EC | |
| miR-152 [70] | Impacts RICTOR and the mTORC2-AKT cascade | Downregulation enables CpG island hypermethylation | |
| Cervical cancer | miR-155 family [71,72] | 3′UTR of PDK1 | Enables PDK1 the activation of the mTOR pathway when under expression |
| miR-99 family [73] | PI3K through the direct target IGF/IGFR | Upregulation directly negatively regulates mTOR expression | |
| miR-634 [74] | Direct binding to 3ÚTR of mTOR | Upregulation represses mTOR expression | |
| miR-338 [13] | No direct information—downregulation of PI3K and AKT, upregulation of pmTOR and p70S6 | Downregulation inhibits autophagy by targeting ATF2 | |
| Ovarian cancer | miR-206 [75] | c-Met | Downregulation impacts estrogen receptor as a direct target, enables growth and invasion in EOC |
| miR-130a [76] | TSC1 | Downregulation enables enhanced mTOR activity | |
| miR-199a [77] | mTOR | Upregulation blocks mTOR expression | |
| miR-100 [78] | FRAP1/mTOR | Down regulation leads to enhanced mTOR pathway activity | |
| MiR-206 [75] | c-Met | Direct activation of downstream AKT/mTOR signaling pathway |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobočan, M.; Bračič, S.; Knez, J.; Takač, I.; Haybaeck, J. The Communication between the PI3K/AKT/mTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer. Cancers 2020, 12, 205. https://doi.org/10.3390/cancers12010205
Sobočan M, Bračič S, Knez J, Takač I, Haybaeck J. The Communication between the PI3K/AKT/mTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer. Cancers. 2020; 12(1):205. https://doi.org/10.3390/cancers12010205
Chicago/Turabian StyleSobočan, Monika, Suzana Bračič, Jure Knez, Iztok Takač, and Johannes Haybaeck. 2020. "The Communication between the PI3K/AKT/mTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer" Cancers 12, no. 1: 205. https://doi.org/10.3390/cancers12010205
APA StyleSobočan, M., Bračič, S., Knez, J., Takač, I., & Haybaeck, J. (2020). The Communication between the PI3K/AKT/mTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer. Cancers, 12(1), 205. https://doi.org/10.3390/cancers12010205