Vimentin Intermediate Filaments as Potential Target for Cancer Treatment

 , , ,
, , ,
Abstract
1. Introduction
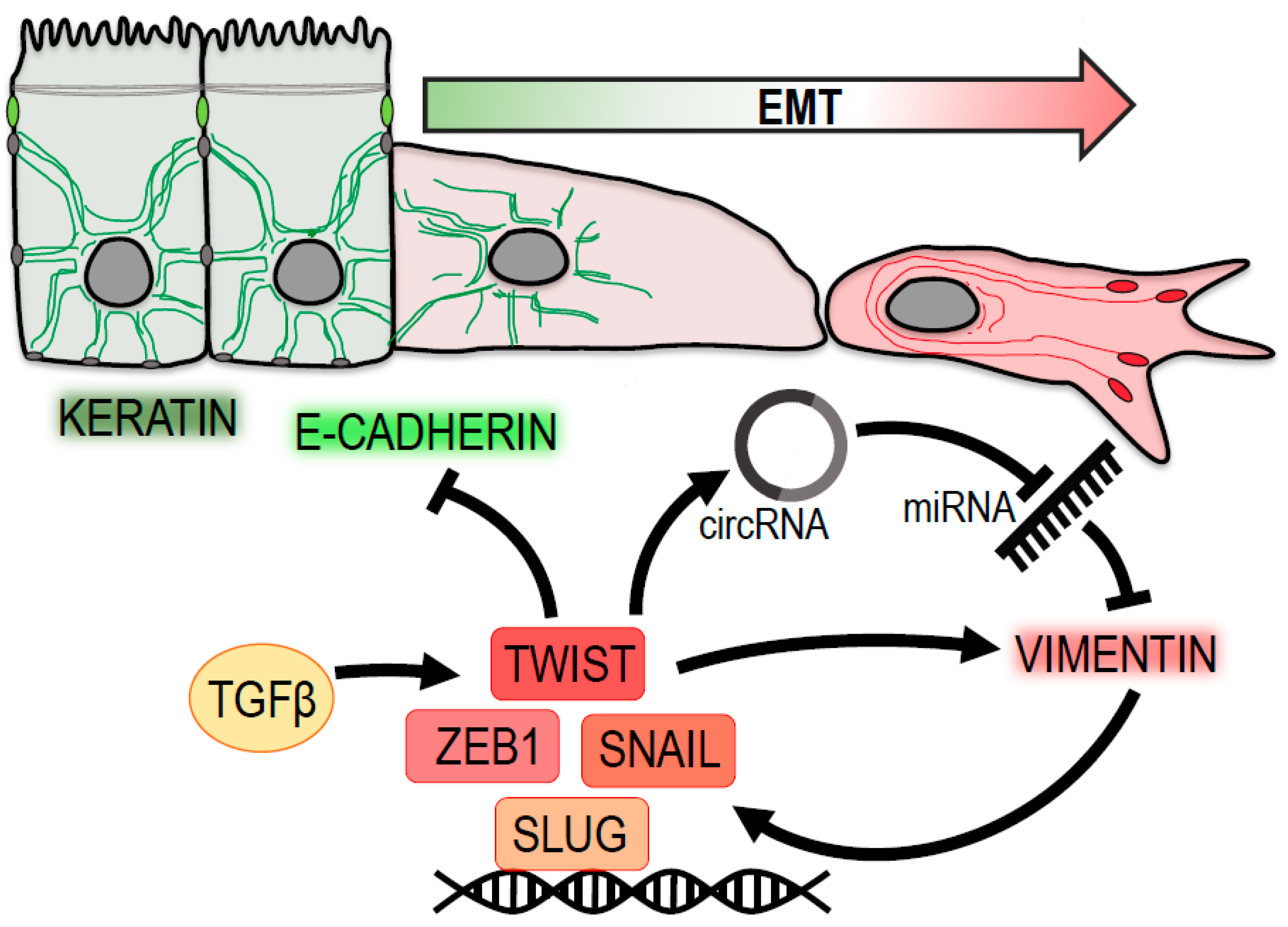
2. Vimentin in EMT
3. Vimentin in the Context of the Cytoskeleton
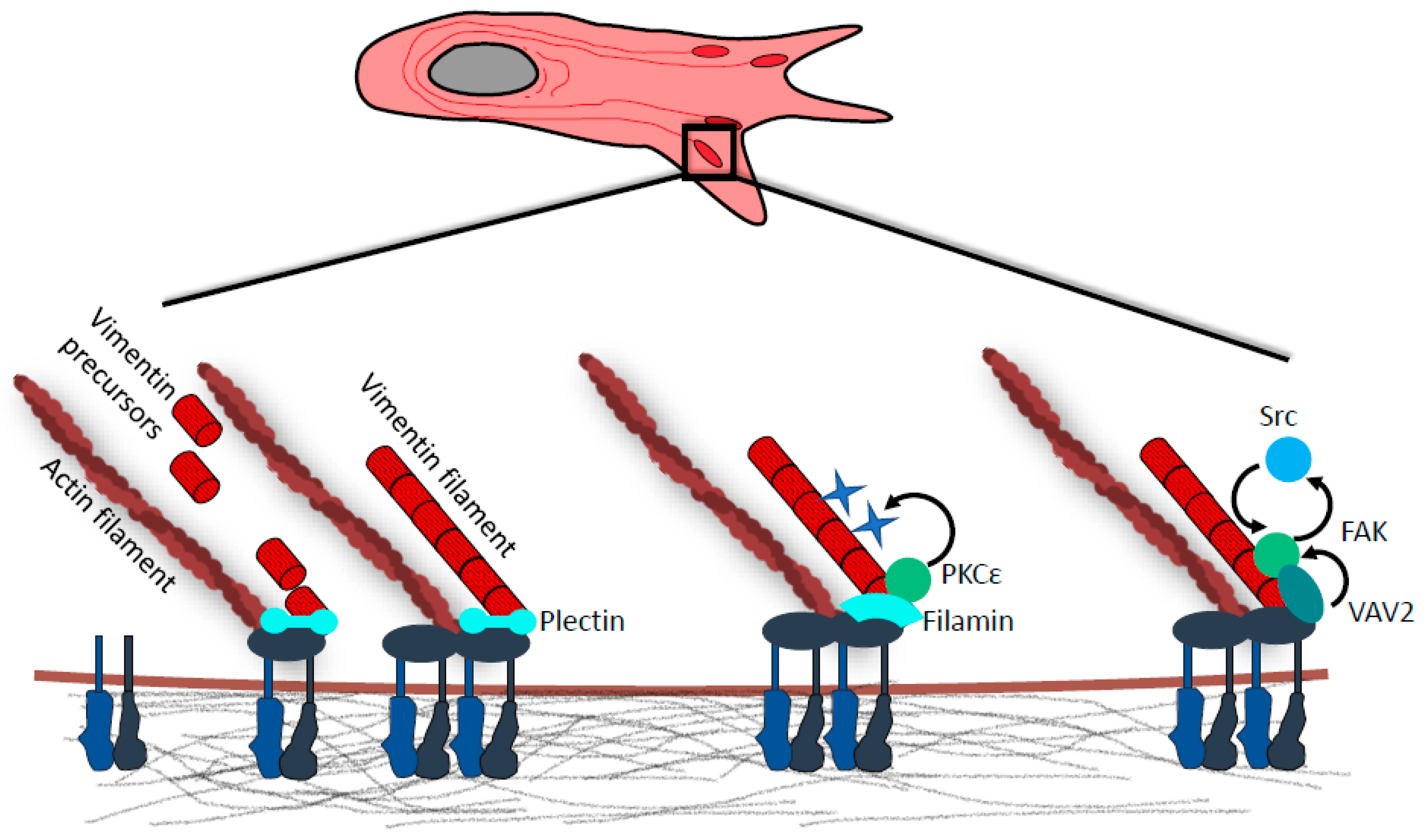
4. Vimentin in Focal Adhesions
5. Vimentin in Migration and Invasion
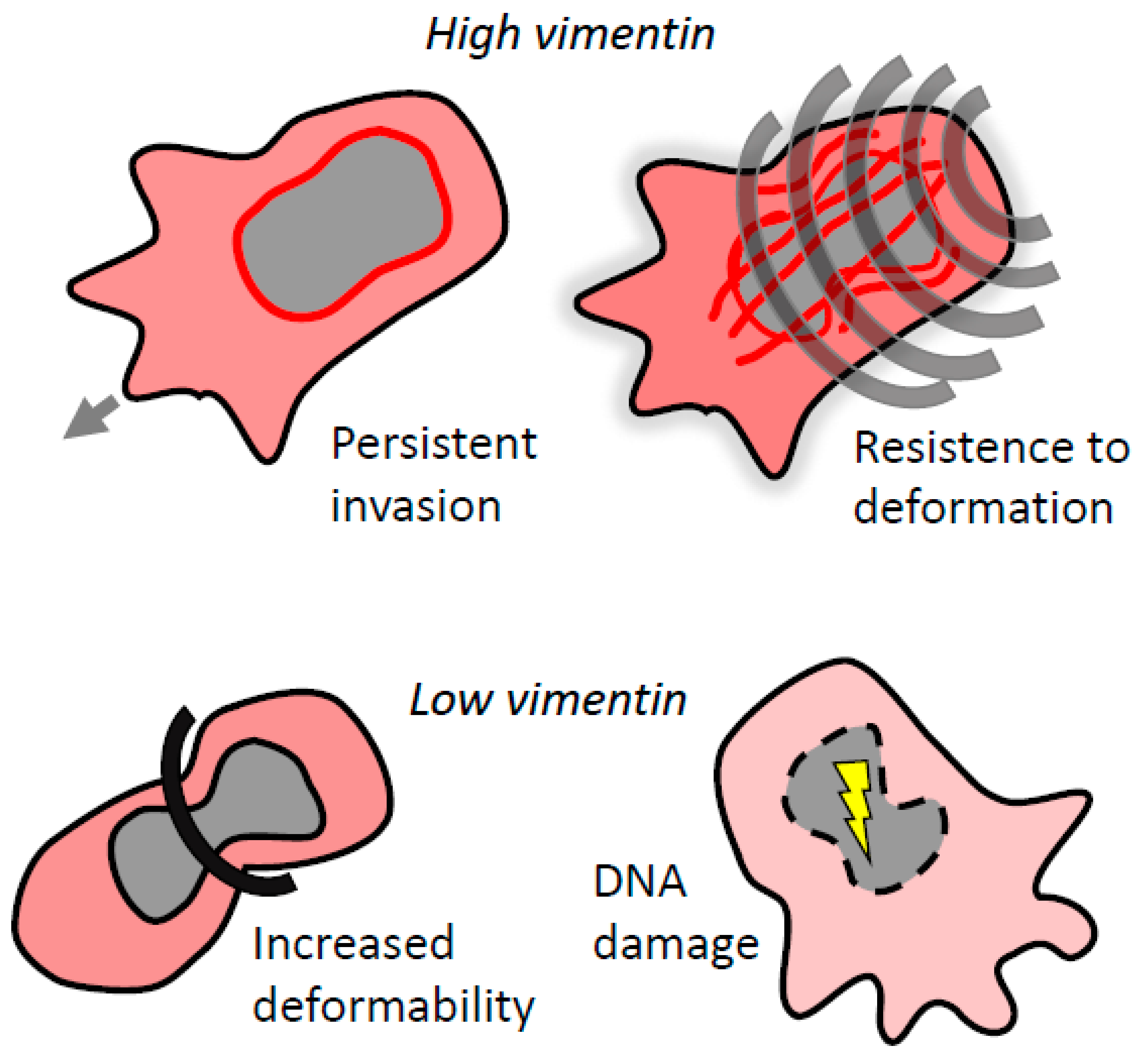
6. Vimentin and the Mechanics of 3D Invasion
7. Research Focused on Vimentin in Metastases
8. Vimentin as a Drug Target
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations:
| Akt | Akt kinase a.k.a. Protein kinase B |
| AP-1 | Activator protein 1 |
| APC | Adenomatous polyposis coli |
| CAFs | Cancer-associated fibroblasts |
| CSC | Cancer stem cell |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal–regulated kinase |
| FA | Focal adhesion |
| FAK | Focal adhesion kinase |
| FOXO3A | Forkhead box O3 |
| GEF | Guanine nucleotide exchange factor |
| GSK | Glycogen synthase kinase |
| HDAC | Histone deacetylase |
| HIF-1 | Hypoxia-inducible factor |
| IFs | Intermediate filaments |
| ILK | Integrin-linked kinase |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen-activated protein kinase |
| MEFs | Mouse embryonic fibroblast |
| MET | Mesenchymal–epithelial transition |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| p38 | p38 MAPK |
| PKC | Protein kinase C |
| Plk | Polo-like kinase |
| RACK1 | Receptor for activated C kinase 1 |
| ROCK | Rho-associated protein kinase |
| S1P | Sphingosine-1-phosphate |
| SHP2 | Protein-tyrosine phosphatase shp2 |
| Snail | Zinc finger transcription repressor Snail |
| SPC | Sphingosylphosphorylcholine |
| Src | Protein-tyrosine kinase Src |
| STAT | Signal transducer and activator of transcription |
| STED | Stimulated emission depletion |
| TCF | Transcription factor TCF |
| TGF-β | Transforming growth factor beta |
| Twist | Zinc finger transcription repressor Twist |
| VAV2 | Guanine nucleotide exchange factor VAV2 |
| WFA | Withaferin A |
| ZEB | Zinc finger E-box-binding homeobox |
References
- Ishikawa, H.; Bischoff, R.; Holtzer, H. Mitosis and intermediate-sized filaments in developing skeletal muscle. J. Cell Boil. 1968, 38, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Magin, T.M.; Weber, K. Genes for intermediate filament proteins and the draft sequence of the human genome: Novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J. Cell Sci. 2001, 114, 2569–2575. [Google Scholar] [PubMed]
- Peter, A.; Stick, R. Evolutionary aspects in intermediate filament proteins. Curr. Opin. Cell Biol. 2015, 32, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef]
- Leduc, C.; Etienne-Manneville, S. Intermediate filaments in cell migration and invasion: The unusual suspects. Curr. Opin. Cell Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Cheng, F.; Eriksson, J.E. Intermediate Filaments and the Regulation of Cell Motility during Regeneration and Wound Healing. Cold Spring Harb. Perspect. Biol. 2017, 9, a022046. [Google Scholar] [CrossRef]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Kokkinos, M.I.; Wafai, R.; Wong, M.K.; Newgreen, D.F.; Thompson, E.W.; Waltham, M. Vimentin and epithelial-mesenchymal transition in human breast cancer—Observations in vitro and in vivo. Cells Tissues Organs 2007, 185, 191–203. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Messica, Y.; Laser-Azogui, A.; Volberg, T.; Elisha, Y.; Lysakovskaia, K.; Eils, R.; Gladilin, E.; Geiger, B.; Beck, R. The role of Vimentin in Regulating Cell Invasive Migration in Dense Cultures of Breast Carcinoma Cells. Nano Lett. 2017, 17, 6941–6948. [Google Scholar] [CrossRef] [PubMed]
- Paccione, R.J.; Miyazaki, H.; Patel, V.; Waseem, A.; Gutkind, J.S.; Zehner, Z.E.; Yeudall, W.A. Keratin down-regulation in vimentin-positive cancer cells is reversible by vimentin RNA interference, which inhibits growth and motility. Mol. Cancer Ther. 2008, 7, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kanamori, Y.; Asano, H.; Hashimoto, O.; Murakami, M.; Kawada, T.; Matsui, T.; Funaba, M. Regulation of brown adipogenesis by the Tgf-beta family: Involvement of Srebp1c in Tgf-beta- and Activin-induced inhibition of adipogenesis. Biochim. Biophys. Acta 2013, 1830, 5027–5035. [Google Scholar] [CrossRef]
- Liu, S.; Liu, L.; Ye, W.; Ye, D.; Wang, T.; Guo, W.; Liao, Y.; Xu, D.; Song, H.; Zhang, L.; et al. High Vimentin Expression Associated with Lymph Node Metastasis and Predicated a Poor Prognosis in Oral Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 38834. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noel, A.; van Roy, F.; Berx, G.; Foidart, J.M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin-ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Salmon, M.; Lin, X.; Zehner, Z.E. TGFbeta1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP-1 and Sp1 family members. Biochim. Biophys. Acta 2007, 1773, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Lilienbaum, A.; Duc Dodon, M.; Alexandre, C.; Gazzolo, L.; Paulin, D. Effect of human T-cell leukemia virus type I tax protein on activation of the human vimentin gene. J. Virol. 1990, 64, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Rittling, S.R.; Coutinho, L.; Amram, T.; Kolbe, M. AP-1/jun binding sites mediate serum inducibility of the human vimentin promoter. Nucleic Acids Res. 1989, 17, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.M. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 2003, 63, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Yao, R.Y.; Sun, Z.Q.; Qiu, W.S.; Yao, Y.S.; Feng, T.T.; Xin, C.; Liang, J.; Yue, L.U. DNA hypermethylation of the vimentin gene inversely correlates with vimentin expression in intestinal- and diffuse-type gastric cancer. Oncol. Lett. 2016, 11, 842–848. [Google Scholar] [CrossRef]
- Zhu, S.; He, C.; Deng, S.; Li, X.; Cui, S.; Zeng, Z.; Liu, M.; Zhao, S.; Chen, J.; Jin, Y.; et al. MiR-548an, Transcriptionally Downregulated by HIF1alpha/HDAC1, Suppresses Tumorigenesis of Pancreatic Cancer by Targeting Vimentin Expression. Mol. Cancer Ther. 2016, 15, 2209–2219. [Google Scholar] [CrossRef]
- Xu, M.; Li, J.; Wang, X.; Meng, S.; Shen, J.; Wang, S.; Xu, X.; Xie, B.; Liu, B.; Xie, L. MiR-22 suppresses epithelial-mesenchymal transition in bladder cancer by inhibiting Snail and MAPK1/Slug/vimentin feedback loop. Cell Death Dis. 2018, 9, 209. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Feng, Z.; Mao, J.; Zhang, C.; Lu, Y.; Li, J.; Zhang, Q.; Li, Q.; Li, L. MicroRNA-138 modulates metastasis and EMT in breast cancer cells by targeting vimentin. Biomed. Pharmacother. 2016, 77, 135–141. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Han, J.X.; Qian, B.; Wang, X.R.; Zhong, W.L.; Qin, Y.; Zhang, H.; Gao, W.F.; Lei, Y.Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindstrom, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef]
- Dmello, C.; Sawant, S.; Alam, H.; Gangadaran, P.; Tiwari, R.; Dongre, H.; Rana, N.; Barve, S.; Costea, D.E.; Chaukar, D.; et al. Vimentin-mediated regulation of cell motility through modulation of beta4 integrin protein levels in oral tumor derived cells. Int. J. Biochem. Cell Biol. 2016, 70, 161–172. [Google Scholar] [CrossRef]
- Leung, C.L.; Green, K.J.; Liem, R.K. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002, 12, 37–45. [Google Scholar] [CrossRef]
- Bouameur, J.E.; Favre, B.; Borradori, L. Plakins, a versatile family of cytolinkers: Roles in skin integrity and in human diseases. J. Investig. Dermatol. 2014, 134, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Osmanagic-Myers, S.; Castanon, M.J. Networking and anchoring through plectin: A key to IF functionality and mechanotransduction. Curr. Opin. Cell Boil. 2015, 32, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bornslaeger, E.A.; Corcoran, C.M.; Stappenbeck, T.S.; Green, K.J. Breaking the connection: Displacement of the desmosomal plaque protein desmoplakin from cell-cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J. Cell Biol. 1996, 134, 985–1001. [Google Scholar] [CrossRef]
- Guo, L.; Degenstein, L.; Dowling, J.; Yu, Q.C.; Wollmann, R.; Perman, B.; Fuchs, E. Gene targeting of BPAG1: Abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 1995, 81, 233–243. [Google Scholar] [CrossRef]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the vimentin network under control: Cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Boil. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Castanon, M.J.; Walko, G.; Winter, L.; Wiche, G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem. Cell Biol. 2013, 140, 33–53. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Boeda, B.; Etienne-Manneville, S. APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 2013, 200, 249–258. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Verkhovsky, A.B.; Borisy, G.G. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996, 135, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263.e258. [Google Scholar] [CrossRef] [PubMed]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Boil. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef] [PubMed]
- Fontao, L.; Geerts, D.; Kuikman, I.; Koster, J.; Kramer, D.; Sonnenberg, A. The interaction of plectin with actin: Evidence for cross-linking of actin filaments by dimerization of the actin-binding domain of plectin. J. Cell Sci. 2001, 114, 2065–2076. [Google Scholar]
- Jiu, Y.; Lehtimaki, J.; Tojkander, S.; Cheng, F.; Jaalinoja, H.; Liu, X.; Varjosalo, M.; Eriksson, J.E.; Lappalainen, P. Bidirectional Interplay between Vimentin Intermediate Filaments and Contractile Actin Stress Fibers. Cell Rep. 2015, 11, 1511–1518. [Google Scholar] [CrossRef]
- Costigliola, N.; Ding, L.; Burckhardt, C.J.; Han, S.J.; Gutierrez, E.; Mota, A.; Groisman, A.; Mitchison, T.J.; Danuser, G. Vimentin fibers orient traction stress. Proc. Natl. Acad. Sci. USA 2017, 114, 5195–5200. [Google Scholar] [CrossRef]
- Jiu, Y.; Peranen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jorgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014, 28, 715–729. [Google Scholar] [CrossRef]
- Walko, G.; Vukasinovic, N.; Gross, K.; Fischer, I.; Sibitz, S.; Fuchs, P.; Reipert, S.; Jungwirth, U.; Berger, W.; Salzer, U.; et al. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011, 7, e1002396. [Google Scholar] [CrossRef]
- Tsuruta, D.; Jones, J.C. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003, 116, 4977–4984. [Google Scholar] [CrossRef]
- Seltmann, K.; Cheng, F.; Wiche, G.; Eriksson, J.E.; Magin, T.M. Keratins Stabilize Hemidesmosomes through Regulation of beta4-Integrin Turnover. J. Investig. Dermatol. 2015, 135, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Kostan, J.; Gregor, M.; Walko, G.; Wiche, G. Plectin Isoform-dependent Regulation of Keratin-Integrin {alpha} 6 {beta} 4 Anchorage via Ca2+/Calmodulin. J. Biol. Chem. 2009, 284, 18525–18536. [Google Scholar] [CrossRef] [PubMed]
- Jirouskova, M.; Nepomucka, K.; Oyman-Eyrilmez, G.; Kalendova, A.; Havelkova, H.; Sarnova, L.; Chalupsky, K.; Schuster, B.; Benada, O.; Miksatkova, P.; et al. Plectin controls biliary tree architecture and stability in cholestasis. J. Hepatol. 2018, 68, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Janostiak, R.; Pataki, A.C.; Brabek, J.; Rosel, D. Mechanosensors in integrin signaling: The emerging role of p130Cas. Eur. J. Cell Biol. 2014, 93, 445–454. [Google Scholar] [CrossRef]
- Kreis, S.; Schonfeld, H.J.; Melchior, C.; Steiner, B.; Kieffer, N. The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp. Cell Res. 2005, 305, 110–121. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Gonzalez, A.M.; Debiase, P.J.; Trejo, H.E.; Goldman, R.D.; Flitney, F.W.; Jones, J.C. Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength. J. Cell Sci. 2009, 122, 1390–1400. [Google Scholar] [CrossRef]
- Kim, H.; Nakamura, F.; Lee, W.; Hong, C.; Perez-Sala, D.; McCulloch, C.A. Regulation of cell adhesion to collagen via beta1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C epsilon. Exp. Cell Res. 2010, 316, 1829–1844. [Google Scholar] [CrossRef]
- Terriac, E.; Coceano, G.; Mavajian, Z.; Hageman, T.A.; Christ, A.F.; Testa, I.; Lautenschlager, F.; Gad, A.K. Vimentin Levels and Serine 71 Phosphorylation in the Control of Cell-Matrix Adhesions, Migration Speed, and Shape of Transformed Human Fibroblasts. Cells 2017, 6, 2. [Google Scholar] [CrossRef]
- Lynch, C.D.; Lazar, A.M.; Iskratsch, T.; Zhang, X.; Sheetz, M.P. Endoplasmic spreading requires coalescence of vimentin intermediate filaments at force-bearing adhesions. Mol. Boil. Cell 2013, 24, 21–30. [Google Scholar] [CrossRef]
- Spurny, R.; Gregor, M.; Castañón, M.J.; Wiche, G. Plectin deficiency affects precursor formation and dynamics of vimentin networks. Exp. Cell Res. 2008, 314, 3570–3580. [Google Scholar] [CrossRef]
- Ivaska, J.; Vuoriluoto, K.; Huovinen, T.; Izawa, I.; Inagaki, M.; Parker, P.J. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005, 24, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, C.; Kim, E.J.; Jang, J.; Kim, S.J.; Kang, S.M.; Kim, M.G.; Jung, H.; Park, D.; Kim, C. Vimentin filaments regulate integrin-ligand interactions by binding to the cytoplasmic tail of integrin beta3. J. Cell Sci. 2016, 129, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Vohnoutka, R.B.; Gulvady, A.C.; Goreczny, G.; Alpha, K.; Handelman, S.K.; Sexton, J.Z.; Turner, C.E. The Focal Adhesion Scaffold Protein Hic-5 Regulates Vimentin Organization in Fibroblasts. Mol. Biol. Cell 2019, 30, 3037–3056. [Google Scholar] [CrossRef] [PubMed]
- Havel, L.S.; Kline, E.R.; Salgueiro, A.M.; Marcus, A.I. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 2015, 34, 1979–1990. [Google Scholar] [CrossRef]
- De Pascalis, C.; Perez-Gonzalez, C.; Seetharaman, S.; Boeda, B.; Vianay, B.; Burute, M.; Leduc, C.; Borghi, N.; Trepat, X.; Etienne-Manneville, S. Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. J. Cell Biol. 2018, 217, 3031–3044. [Google Scholar] [CrossRef]
- Leube, R.E.; Moch, M.; Windoffer, R. Intermediate filaments and the regulation of focal adhesion. Curr. Opin. Cell Boil. 2015, 32, 13–20. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Wiche, G. Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: A novel mechanism to regulate protein kinase C activity. J. Biol. Chem. 2004, 279, 18701–18710. [Google Scholar] [CrossRef]
- Dave, J.M.; Kang, H.; Abbey, C.A.; Maxwell, S.A.; Bayless, K.J. Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK). J. Biol. Chem. 2013, 288, 30720–30733. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Gregor, M.; Walko, G.; Burgstaller, G.; Reipert, S.; Wiche, G. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 2006, 174, 557–568. [Google Scholar] [CrossRef]
- Murray, M.E.; Mendez, M.G.; Janmey, P.A. Substrate stiffness regulates solubility of cellular vimentin. Mol. Biol. Cell 2014, 25, 87–94. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Zahm, J.M.; Tournier, J.M.; Volders, L.; Foidart, J.M.; Birembaut, P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 1999, 112 Pt 24, 4615–4625. [Google Scholar]
- Rodriguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; Lopez, L.; Serrano, S.; de Herreros, A.G.; Rodriguez-Manzaneque, J.C.; et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Eckes, B.; Colucci-Guyon, E.; Smola, H.; Nodder, S.; Babinet, C.; Krieg, T.; Martin, P. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 2000, 113 Pt 13, 2455–2462. [Google Scholar]
- Yoshida, K.; Saito, T.; Kamida, A.; Matsumoto, K.; Saeki, K.; Mochizuki, M.; Sasaki, N.; Nakagawa, T. Transforming growth factor-beta transiently induces vimentin expression and invasive capacity in a canine mammary gland tumor cell line. Res. Vet. Sci. 2013, 94, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Osorio, L.A.; Farfan, N.M.; Castellon, E.A.; Contreras, H.R. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol. Med. Rep. 2016, 13, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chang, P.W.; Hsu, W.H.; Chang, H.C.; Chen, C.L.; Lai, C.C.; Chiu, W.T.; Chen, H.C. Src and SHP2 coordinately regulate the dynamics and organization of vimentin filaments during cell migration. Oncogene 2019, 38, 4075–4094. [Google Scholar] [CrossRef]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef]
- Colburn, Z.T.; Jones, J.C.R. Complexes of alpha6beta4 integrin and vimentin act as signaling hubs to regulate epithelial cell migration. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Pankova, K.; Rosel, D.; Novotny, M.; Brabek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol. Life Sci. 2010, 67, 63–71. [Google Scholar] [CrossRef]
- Mierke, C.T.; Rosel, D.; Fabry, B.; Brabek, J. Contractile forces in tumor cell migration. Eur. J. Cell Biol. 2008, 87, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Koster, S.; Weitz, D.A.; Goldman, R.D.; Aebi, U.; Herrmann, H. Intermediate filament mechanics in vitro and in the cell: From coiled coils to filaments, fibers and networks. Curr. Opin. Cell Boil. 2015, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Rathje, L.S.; Nordgren, N.; Pettersson, T.; Ronnlund, D.; Widengren, J.; Aspenstrom, P.; Gad, A.K. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef]
- Petrie, R.J.; Koo, H.; Yamada, K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 2014, 345, 1062–1065. [Google Scholar] [CrossRef]
- Stankevicins, L.D.C.; Urbanska, M.; Flormann, D.A.; Terriac, E.; Mostajeran, Z.; Gad, A.K.B.; Cheng, F.; Eriksson, J.E.; Lautenschläger, F. Vimentin provides the mechanical resilience required for amoeboid migration and protection of the nucleus. bioRxiv 2019. [Google Scholar] [CrossRef]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef]
- Tudor, S.M.; Lavenus, S.B.; Logue, J.S. A flexible network of Vimentin intermediate filaments promotes the migration of amoeboid cancer cells through confined environments. bioRxiv 2019. [Google Scholar] [CrossRef]
- Terriac, E.; Schutz, S.; Lautenschlager, F. Vimentin Intermediate Filament Rings Deform the Nucleus during the First Steps of Adhesion. Front. Cell Dev. Biol. 2019, 7, 106. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rosse, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Sutoh Yoneyama, M.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.I.; Kang, H.; Dave, J.M.; Mendoza, E.A.; Su, S.C.; Maxwell, S.A.; Bayless, K.J. Calpain-mediated vimentin cleavage occurs upstream of MT1-MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis 2012, 15, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Hyder, C.L.; Kemppainen, K.; Isoniemi, K.O.; Imanishi, S.Y.; Goto, H.; Inagaki, M.; Fazeli, E.; Eriksson, J.E.; Tornquist, K. Sphingolipids inhibit vimentin-dependent cell migration. J. Cell Sci. 2015, 128, 2057–2069. [Google Scholar] [CrossRef]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.T.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Sleeman, J.; Steeg, P.S. Cancer metastasis as a therapeutic target. Eur. J. Cancer 2010, 46, 1177–1180. [Google Scholar] [CrossRef]
- Gandalovicova, A.; Rosel, D.; Fernandes, M.; Vesely, P.; Heneberg, P.; Cermak, V.; Petruzelka, L.; Kumar, S.; Sanz-Moreno, V.; Brabek, J. Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Kaschula, C.H.; Tuveri, R.; Ngarande, E.; Dzobo, K.; Barnett, C.; Kusza, D.A.; Graham, L.M.; Katz, A.A.; Rafudeen, M.S.; Parker, M.I.; et al. The garlic compound ajoene covalently binds vimentin, disrupts the vimentin network and exerts anti-metastatic activity in cancer cells. BMC Cancer 2019, 19, 248. [Google Scholar] [CrossRef]
- Burikhanov, R.; Sviripa, V.M.; Hebbar, N.; Zhang, W.; Layton, W.J.; Hamza, A.; Zhan, C.G.; Watt, D.S.; Liu, C.; Rangnekar, V.M. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nat. Chem. Biol. 2014, 10, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhou, S.; Zhan, Y.; Ke, J.; Wang, K.; Liang, Q.; Hou, Y.; Zhu, P.; Ao, W.; Wei, X.; et al. Dioscin Inhibits the Invasion and Migration of Hepatocellular Carcinoma HepG2 Cells by Reversing TGF-beta1-Induced Epithelial-Mesenchymal Transition. Molecules 2019, 24, 2222. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Pietila, M.; Pearson, A.D.; Sarkar, T.R.; Ahmad, I.; Soundararajan, R.; Lyssiotis, C.A.; Mani, S.A.; Schultz, P.G.; Lairson, L.L. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc. Natl. Acad. Sci. USA 2017, 114, E9903–E9912. [Google Scholar] [CrossRef] [PubMed]
- Kanugula, A.K.; Dhople, V.M.; Volker, U.; Ummanni, R.; Kotamraju, S. Fluvastatin mediated breast cancer cell death: A proteomic approach to identify differentially regulated proteins in MDA-MB-231 cells. PLoS ONE 2014, 9, e108890. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, W.I.; Jeon, B.N.; Choi, K.C.; Kim, K.; Kim, T.J.; Ham, J.; Jang, H.J.; Kang, K.S.; Ko, H. Stereospecific effects of ginsenoside 20-Rg3 inhibits TGF-beta1-induced epithelial-mesenchymal transition and suppresses lung cancer migration, invasion and anoikis resistance. Toxicology 2014, 322, 23–33. [Google Scholar] [CrossRef]
- Zamay, T.N.; Kolovskaya, O.S.; Glazyrin, Y.E.; Zamay, G.S.; Kuznetsova, S.A.; Spivak, E.A.; Wehbe, M.; Savitskaya, A.G.; Zubkova, O.A.; Kadkina, A.; et al. DNA-aptamer targeting vimentin for tumor therapy in vivo. Nucleic Acid Ther. 2014, 24, 160–170. [Google Scholar] [CrossRef]
- Yoon, S.; Armstrong, B.; Habib, N.; Rossi, J.J. Blind SELEX Approach Identifies RNA Aptamers That Regulate EMT and Inhibit Metastasis. Mol. Cancer Res. 2017, 15, 811–820. [Google Scholar] [CrossRef]
- Ji, Q.; Liu, X.; Han, Z.; Zhou, L.; Sui, H.; Yan, L.; Jiang, H.; Ren, J.; Cai, J.; Li, Q. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-beta1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer 2015, 15, 97. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Shibuya, M.; Sugawara, H.; Kawaguchi, O.; Hirsoe, C. Salinomycin, a new polyether antibiotic. J. Antibiot. 1974, 27, 814–821. [Google Scholar] [CrossRef]
- Dong, T.T.; Zhou, H.M.; Wang, L.L.; Feng, B.; Lv, B.; Zheng, M.H. Salinomycin selectively targets ‘CD133+’ cell subpopulations and decreases malignant traits in colorectal cancer lines. Ann. Surg. Oncol. 2011, 18, 1797–1804. [Google Scholar] [CrossRef]
- Li, R.; Dong, T.; Hu, C.; Lu, J.; Dai, J.; Liu, P. Salinomycin repressed the epithelial-mesenchymal transition of epithelial ovarian cancer cells via downregulating Wnt/beta-catenin pathway. Onco Targets Ther. 2017, 10, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Raina, K.; Sharma, G.; Agarwal, R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin. Cancer Res. 2008, 14, 7773–7780. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zeng, J.; Li, L.; Fan, J.; Zhang, D.; Xue, Y.; Zhu, G.; Yang, L.; Wang, X.; He, D. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol. Rep. 2010, 23, 1545–1552. [Google Scholar] [PubMed]
- Trogden, K.P.; Battaglia, R.A.; Kabiraj, P.; Madden, V.J.; Herrmann, H.; Snider, N.T. An image-based small-molecule screen identifies vimentin as a pharmacologically relevant target of simvastatin in cancer cells. FASEB J. 2018, 32, 2841–2854. [Google Scholar] [CrossRef]
- Hajar, R. Statins: Past and present. Heart Views 2011, 12, 121–127. [Google Scholar] [CrossRef]
- Wang, X.; Wang, T.; Yi, F.; Duan, C.; Wanwg, Q.; He, N.; Zhu, L.; Li, Q.; Deng, W. Ursolic Acid Inhibits Tumor Growth via Epithelial-to-Mesenchymal Transition in Colorectal Cancer Cells. Biol. Pharm. Bull. 2019, 42, 685–691. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Singh, R.; Peng, S.; Viswanath, P.; Sambandam, V.; Shen, L.; Rao, X.; Fang, B.; Wang, J.; Johnson, F.M. Non-canonical cMet regulation by vimentin mediates Plk1 inhibitor-induced apoptosis. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Lee, D.H.; Lim, I.H.; Sung, E.G.; Kim, J.Y.; Song, I.H.; Park, Y.K.; Lee, T.J. Withaferin A inhibits matrix metalloproteinase-9 activity by suppressing the Akt signaling pathway. Oncol. Rep. 2013, 30, 933–938. [Google Scholar] [CrossRef]
- Shohat, B.; Gitter, S.; Abraham, A.; Lavie, D. Antitumor activity of withaferin A (NSC-101088). Cancer Chemother. Rep. 1967, 51, 271–276. [Google Scholar]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Yan, J.; Hong, S.; Kong, L.Y.; Gabrusiewicz, K.; Xia, X.; Heimberger, A.B.; Li, S. Discovery of cell surface vimentin targeting mAb for direct disruption of GBM tumor initiating cells. Oncotarget 2016, 7, 72021–72032. [Google Scholar] [CrossRef] [PubMed]
- Bargagna-Mohan, P.; Hamza, A.; Kim, Y.E.; Khuan Abby Ho, Y.; Mor-Vaknin, N.; Wendschlag, N.; Liu, J.; Evans, R.M.; Markovitz, D.M.; Zhan, C.G.; et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007, 14, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; Haegeman, G.; Heyninck, K. Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol. 2012, 84, 1282–1291. [Google Scholar] [CrossRef]
- Stan, S.D.; Hahm, E.R.; Warin, R.; Singh, S.V. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008, 68, 7661–7669. [Google Scholar] [CrossRef]
- Lee, J.; Hahm, E.R.; Marcus, A.I.; Singh, S.V. Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors. Mol. Carcinog. 2015, 54, 417–429. [Google Scholar] [CrossRef]
- Ruan, J.S.; Zhou, H.; Yang, L.; Wang, L.; Jiang, Z.S.; Sun, H.; Wang, S.M. Ursolic Acid Attenuates TGF-beta1-Induced Epithelial-Mesenchymal Transition in NSCLC by Targeting Integrin alphaVbeta5/MMPs Signaling. Oncol. Res. 2019, 27, 593–600. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef]
- Shigyo, M.; Kuboyama, T.; Sawai, Y.; Tada-Umezaki, M.; Tohda, C. Extracellular vimentin interacts with insulin-like growth factor 1 receptor to promote axonal growth. Sci. Rep. 2015, 5, 12055. [Google Scholar] [CrossRef]
- Sun, S.; Poon, R.T.; Lee, N.P.; Yeung, C.; Chan, K.L.; Ng, I.O.; Day, P.J.; Luk, J.M. Proteomics of hepatocellular carcinoma: Serum vimentin as a surrogate marker for small tumors (<or = 2 cm). J. Proteome Res. 2010, 9, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhang, X.; Zhang, Q.; Yang, J.; Chen, Q.; Wang, J.; Li, X.; Chen, J.; Ma, T.; Li, G.; et al. Vimentin-positive circulating tumor cells as a biomarker for diagnosis and treatment monitoring in patients with pancreatic cancer. Cancer Lett. 2019, 452, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Bleaken, B.M.; Romisher, A.R.; Alnwibit, A.A.; Menko, A.S. In wound repair vimentin mediates the transition of mesenchymal leader cells to a myofibroblast phenotype. Mol. Biol. Cell 2018, 29, 1555–1570. [Google Scholar] [CrossRef] [PubMed]
- Haversen, L.; Sundelin, J.P.; Mardinoglu, A.; Rutberg, M.; Stahlman, M.; Wilhelmsson, U.; Hulten, L.M.; Pekny, M.; Fogelstrand, P.; Bentzon, J.F.; et al. Vimentin deficiency in macrophages induces increased oxidative stress and vascular inflammation but attenuates atherosclerosis in mice. Sci. Rep. 2018, 8, 16973. [Google Scholar] [CrossRef]
- Colucci-Guyon, E.; Portier, M.M.; Dunia, I.; Paulin, D.; Pournin, S.; Babinet, C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell 1994, 79, 679–694. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Mode of Action | Effect | Direct Binding | Origin | Cancer-Related Clinical Trials * | References |
|---|---|---|---|---|---|---|
| Aojene | Vimentin IF collapse | Anti-invasive/migratory | Yes | Allium sativum (garlic) | - | [100] |
| Arylquin 1 | Par-4 displacement from vimentin, Par-4 secretion | Paracrine apoptosis induction in cancer cells | Yes | 3-arylquinoline derivative | [101] | |
| Dioscin | Suppression of vimentin expression via TGF-β1 pathway | EMT reversal, anti-invasive/migratory | NK | steroid saponin isolated from Chinese medicinal plants | - | [102] |
| FiVe1 | Ser56 phosphorylation, vimentin IF collapse | MET, mitotic catastrophe in vimentin-containing cells | Yes | Unknown | - | [103] |
| Fluvastatin | Caspase-3-mediated proteolysis of vimentin | Cytotoxicity in invasive cancer cells | NK | synthetic indole-heptanoic acid derivative | Phase 1-2 | [104] |
| Ginsenoside 20(R)-Rg3 | Suppression of vimentin expression via TGF-β1 pathway | EMT suppression | NK | Panax ginseng (ginseng) | Phase 2 | [105] |
| NAS-24 | Unknown | Apoptosis induction in cancer cells | Yes | synthetic | - | [106] |
| P15 | Unknown | Anti-invasive/migratory | Yes | synthetic | - | [107] |
| Resveratrol | Suppression of vimentin expression via TGF-β1 pathway | Anti-invasive/migratory, metastasis reduction | NK | phytoalexin found in red grapes and other plants | Phase 1-2 | [108] |
| Salinomycin | Suppression of vimentin expression via Wnt/β-catenin | MET, anti-migratory, CSC reduction | NK | Streptomyces albus | - | [109,110,111] |
| Silibinin | Suppression of vimentin expression | EMT suppression, anti-invasive/migratory, tumor dissemination and growth reduction | NK | Silybum marianum | Phase 1-2 | [112,113] |
| Simvastatin | Vimentin IF collapse | Apoptosis induction in vimentin-containing cells | NK | derivative of afermentation product of Aspergillus terreus | Phase 1-3 (FDA approved for Type II diabetes) | [114,115] |
| Ursolic acid | Suppression of vimentin expression possibly via TGF-β1 pathway | Anti-invasive/migratory, tumor growth reduction, apoptosis | NK | pentacyclic triterpenic acid found in plants | - | [116] |
| Volasertib | Decrease in Ser82 phosphorylation caused by Plk1 inhibition, loss of cMet phosphorylation via β1-integrin | MET, anti-invasive/migratory, apoptosis | NK | dihydropteridinone derivative | Phase 1-3 | [117,118] |
| Withaferin A | Doses below 500 nM: Ser56 phosphorylation, vimentin IF collapse Higher doses: vimentin expression lowered | Dose below 500 nM: anti-invasive/migratory in cancer cells Higher doses: MET, apoptosis induction (vimentin independent) | Yes | Withania somnifera | - | [119,120,121] |
| 86C | Internalization of cell surface vimentin, otherwise unknown | Apoptosis induction, tumor growth reduction | Yes | monoclonal antibody | Phase 1 | [122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strouhalova, K.; Přechová, M.; Gandalovičová, A.; Brábek, J.; Gregor, M.; Rosel, D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers 2020, 12, 184. https://doi.org/10.3390/cancers12010184
Strouhalova K, Přechová M, Gandalovičová A, Brábek J, Gregor M, Rosel D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers. 2020; 12(1):184. https://doi.org/10.3390/cancers12010184
Chicago/Turabian StyleStrouhalova, Katerina, Magdalena Přechová, Aneta Gandalovičová, Jan Brábek, Martin Gregor, and Daniel Rosel. 2020. "Vimentin Intermediate Filaments as Potential Target for Cancer Treatment" Cancers 12, no. 1: 184. https://doi.org/10.3390/cancers12010184
APA StyleStrouhalova, K., Přechová, M., Gandalovičová, A., Brábek, J., Gregor, M., & Rosel, D. (2020). Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers, 12(1), 184. https://doi.org/10.3390/cancers12010184