Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles

 ,
,  , ,
, ,  ,
, 
Abstract
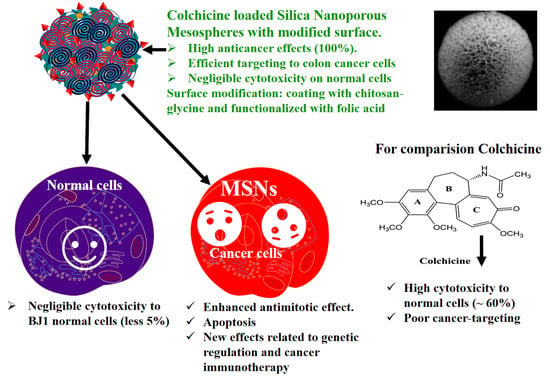
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of the Targeted Delivery System
2.2. Electron Microscopy of Materials
2.3. Surface Area Characteristics
2.4. Elemental Content Analysis
2.5. Particle Size Measurement
2.6. Functional Group Determination
2.7. Simultaneous Thermal Analysis (STA-DSC) and XRD Characterization
2.8. Zeta Potential Measurements
2.9. MSN Cytotoxicity Evaluation
2.10. In Vitro Anticancer Effects against Cancer Cells
2.11. Inhibition of Tubulin Activity in HCT116 Cancer Cells
2.12. Cell Cycle Arrest at the G2/M Phase in HCT116 Cancer Cells
2.13. Enhancement of Apoptosis Induction in HCT116 Cancer Cells
2.14. Activation of Caspase-3 in HCT116 Cancer Cells
2.15. Modulation of Proapoptotic Bax/Bcl-2 in HCT116 Cancer Cells
2.16. Inhibition of BRAF Expression in HCT116 Cancer Cells
2.17. Enhancement of Cytochrome c Triggers in HCT116 Cancer Cells
2.18. Reduction of Mitochondrial Membrane Potential in HCT116 Cancer Cells
2.19. Inhibition of CD44 Expression in HCT116 Cancer Cells
2.20. Inhibition of MALAT-1 Expression in HCT116 Cancer Cells
2.21. Attenuation of mir-205 Expression in HCT116 Cancer Cells
2.22. Inhibition of Ang-2 Expression in HCT116 Cancer Cells
2.23. Inhibition of PD-1 Expression in HCT116 Cancer Cells
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis and Modification of Mesoporous Silica Nanoparticles
3.2.2. Colchicine Loading
3.2.3. Coating and Conjugation with Chitosan-Glycine and Folic Acid
3.2.4. Characterization Techniques
3.2.5. In Vitro Cytotoxicity Assessment
3.2.6. Apoptosis Detection and Cell Cycle Analysis with Flow Cytometry
3.2.7. Caspase-3 Activity Assay
3.2.8. Tubulin Assay
3.2.9. Expression of Bax and Bcl-2
Cell Culture Treatment and RNA Extraction
Quantitative Determination by RT-PCR
3.2.10. BRAF Assay
3.2.11. Cytochrome c Assay
3.2.12. Measurement of Mitochondrial Membrane Potential
3.2.13. Assays for MALAT-1, mir-205, Ang-2-CD44, and PD-1
Cell Culture and Treatment
3.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaplan, M.M.; Alling, D.W.; Zimmerman, H.J.; Wolfe, H.J.; Sepersky, R.A.; Hirsch, G.S.; Elta, G.H.; Glick, K.A.; Eagen, K.A. A prospective trial of colchicine for primary biliary cirrhosis. N. Engl. J. Med. 1986, 315, 1448–1454. [Google Scholar] [CrossRef]
- Cohen, A.S.; Rubinow, A.; Anderson, J.J.; Skinner, M.; Mason, J.H.; Libbey, C.; Kayne, H. Survival of patients with primary (AL) amyloidosis: Colchicine-treated cases from 1976 to 1983 compared with cases seen in previous years (1961 to 1973). Am. J. Med. 1987, 82, 1182–1190. [Google Scholar] [CrossRef]
- Gigax, J.H.; Robison, J.R. The Successful Treatment of Intraurethral Condyloma Acuminata with Colchicine. J. Urol. 1971, 105, 809–811. [Google Scholar] [CrossRef]
- Sampedro-Núñez, M.; Serrano-Somavilla, A.; Adrados, M.; Cameselle-Teijeiro, J.M.; Blanco-Carrera, C.; Cabezas-Agricola, J.M.; Martínez-Hernández, R.; Martín-Pérez, E.; Muñoz de Nova, J.L.; Díaz, J.Á.; et al. Analysis of expression of the PD-1/PD-L1 immune checkpoint system and its prognostic impact in gastroenteropancreatic neuroendocrine tumors. Sci. Rep. 2018, 8, 17812. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, Y.; Peng, W. Colchicine induces apoptosis in HT29 human colon cancer cells via the AKT and c-Jun N-terminal kinase signaling pathways. Mol. Med. Rep. 2015, 12, 5939–5944. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Z.; Granieri, L.; Pasculescu, A.; Datti, A.; Asa, S.L.; Xu, Z.; Ezzat, S. High-throughput drug library screening identifies colchicine as a thyroid cancer inhibitor. Oncotarget 2016, 7, 19948–19959. [Google Scholar] [CrossRef]
- Cho, J.H.; Joo, Y.H.; Shin, E.Y.; Park, E.J.; Kim, M.S. Anticancer Effects of Colchicine on Hypopharyngeal Cancer. Anticancer Res. 2017, 37, 6269–6280. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Kuo, C.H.; Wu, D.C.; Chuang, W.L. Anticancer effects of clinically acceptable colchicine concentrations on human gastric cancer cell lines. Kaohsiung J. Med. Sci. 2016, 32, 68–73. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, P.R.; Mondhe, D.M. Potential anticancer role of colchicine-based derivatives: An overview. Anti Cancer Drugs 2017, 28, 250–262. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, B.; Sharma, P.R.; Bharate, S.B.; Saxena, A.K.; Mondhe, D.M. A novel microtubule depolymerizing colchicine analogue triggers apoptosis and autophagy in HCT-116 colon cancer cells. Cell Biochem. Funct. 2016, 34, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.R.; Kim, J.H.; Cheon, J.H.; Kim, H.S.; Yoon, S. Attenuation of Colchicine Toxicity in Drug-resistant Cancer Cells by Co-treatment with Anti-malarial Drugs. Anticancer Res. 2016, 36, 5859–5866. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Dudani, A.K. Mechanism of action of antimitotic drugs: A new hypothesis based on the role of cellular calcium. Med. Hypotheses 1989, 28, 57–69. [Google Scholar] [CrossRef]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Qi, C.; Wang, X.; Shen, Z.; Chen, S.; Yu, H.; Williams, N.; Wang, G. Anti-mitotic chemotherapeutics promote apoptosis through TL1A-activated death receptor 3 in cancer cells. Cell Res. 2018, 28, 544–555. [Google Scholar] [CrossRef]
- Fong, A.; Durkin, A.; Lee, H. The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. Int. J. Mol. Sci. 2019, 20, 586. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Silva, R.; Gullo, I.; Carneiro, F. The PD-1:PD-L1 immune inhibitory checkpoint in Helicobacter pylori infection and gastric cancer: A comprehensive review and future perspectives. Porto Biomed. J. 2016, 1, 4–11. [Google Scholar] [CrossRef]
- Yao, H.; Wang, H.; Li, C.; Fang, J.-Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef]
- Berntsson, J.; Eberhard, J.; Nodin, B.; Leandersson, K.; Larsson, A.H.; Jirström, K. Expression of programmed cell death protein 1 (PD-1) and its ligand PD-L1 in colorectal cancer: Relationship with sidedness and prognosis. Oncoimmunology 2018, 7, e1465165. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Franchini, D.-M.; Lanvin, O.; Tosolini, M.; Patras de Campaigno, E.; Cammas, A.; Péricart, S.; Scarlata, C.-M.; Lebras, M.; Rossi, C.; Ligat, L.; et al. Microtubule-Driven Stress Granule Dynamics Regulate Inhibitory Immune Checkpoint Expression in T Cells. Cell Rep. 2019, 26, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Ades, S. Adjuvant chemotherapy for colon cancer in the elderly: Moving from evidence to practice. Oncology 2009, 23, 162–167. [Google Scholar] [PubMed]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: Current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- AbouAitah, K.; Swiderska-Sroda, A.; Farghali, A.A.; Wojnarowicz, J.; Stefanek, A.; Gierlotka, S.; Opalinska, A.; Allayeh, A.K.; Ciach, T.; Lojkowski, W. Folic acid-conjugated mesoporous silica particles as nanocarriers of natural prodrugs for cancer targeting and antioxidant action. Oncotarget 2018, 9, 26466–26490. [Google Scholar] [CrossRef] [PubMed]
- Shahein, S.A.; Aboul-Enein, A.M.; Higazy, I.M.; Abou-Elella, F.; Lojkowski, W.; Ahmed, E.R.; Mousa, S.A.; AbouAitah, K. Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release. Int. J. Nanomed. 2019, 14, 5503–5526. [Google Scholar] [CrossRef]
- Cauda, V.; Engelke, H.; Sauer, A.; Arcizet, D.; Brauchle, C.; Radler, J.; Bein, T. Colchicine-loaded lipid bilayer-coated 50 nm mesoporous nanoparticles efficiently induce microtubule depolymerization upon cell uptake. Nano Lett. 2010, 10, 2484–2492. [Google Scholar] [CrossRef]
- Muvaffak, A.; Gurhan, I.; Hasirci, N. Prolonged cytotoxic effect of colchicine released from biodegradable microspheres. J. Biomed. Mater. Res. Part B Appl. Biomater. 2004, 71, 295–304. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Cha, D.; Zhang, X.; Basset, J.M. High-surface-area silica nanospheres (KCC-1) with a fibrous morphology. Angew. Chem. Int. Ed. Engl. 2010, 49, 9652–9656. [Google Scholar] [CrossRef]
- Chang, B.; Chen, D.; Wang, Y.; Chen, Y.; Jiao, Y.; Sha, X.; Yang, W. Bioresponsive Controlled Drug Release Based on Mesoporous Silica Nanoparticles Coated with Reductively Sheddable Polymer Shell. Chem. Mater. 2013, 25, 574–585. [Google Scholar] [CrossRef]
- Castillo, R.R.; Lozano, D.; González, B.; Manzano, M.; Izquierdo-Barba, I.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery: An update. Expert Opin. Drug Deliv. 2019, 16, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef] [PubMed]
- Sabharanjak, S.; Mayor, S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 2004, 56, 1099–1109. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Ali Mansoori, G.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef]
- AbouAitah, K.; Farghali, A.; Swiderska-Sroda, A.; Lojkowski, W.; Razin, A.; Khedr, M.K. Mesoporous silica materials in drug delivery system: pH/glutathione-responsive release of poorly water-soluble pro-drug quercetin from two and three-dimensional pore-structure nanoparticles. J. Nanomed. Nanotechnol. 2016, 7. [Google Scholar] [CrossRef]
- AbouAitah, K.E.; Farghali, A.; Swiderska-Sroda, A.; Lojkowski, W.; Razin, A.; Khedr, M.K. pH-controlled release system for curcumin based on functionalized dendritic mesoporous silica nanoparticles. J. Nanomed. Nanotechnol. 2016, 7. [Google Scholar] [CrossRef]
- Menon, N.; Leong, D.T. Cytotoxic Effects of Phosphonate-Functionalized Mesoporous Silica Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 2416–2422. [Google Scholar] [CrossRef]
- Villanueva, A.; Canete, M.; Roca, A.G.; Calero, M.; Veintemillas-Verdaguer, S.; Serna, C.J.; Morales Mdel, P.; Miranda, R. The influence of surface functionalization on the enhanced internalization of magnetic nanoparticles in cancer cells. Nanotechnology 2009, 20, 115103. [Google Scholar] [CrossRef]
- Yue, Z.-G.; Wei, W.; Lv, P.-P.; Yue, H.; Wang, L.-Y.; Su, Z.-G.; Ma, G.-H. Surface Charge Affects Cellular Uptake and Intracellular Trafficking of Chitosan-Based Nanoparticles. Biomacromolecules 2011, 12, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Yin, M.; Zhao, L.; Meng, F.; Luo, L. Recent progress on nanoparticle-based drug delivery systems for cancer therapy. Cancer Biol. Med. 2017, 14, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef]
- Chen, T.; Wu, W.; Xiao, H.; Chen, Y.; Chen, M.; Li, J. Intelligent Drug Delivery System Based on Mesoporous Silica Nanoparticles Coated with an Ultra-pH-Sensitive Gatekeeper and Poly(ethylene glycol). ACS Macro Lett. 2016, 5, 55–58. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef]
- Andreu, J.M.; Perez-Ramirez, B.; Gorbunoff, M.J.; Ayala, D.; Timasheff, S.N. Role of the colchicine ring A and its methoxy groups in the binding to tubulin and microtubule inhibition. Biochemistry 1998, 37, 8356–8368. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Blajeski, A.L.; Phan, V.A.; Kottke, T.J.; Kaufmann, S.H. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Investig. 2002, 110, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.A.; Patil, R.; Patil, S.A.; Beaman, K.D. Microtubule inhibitor, SP-6-27 inhibits angiogenesis and induces apoptosis in ovarian cancer cells. Oncotarget 2017, 8, 67017–67028. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, A.R.; O’Connell, M.J. Cell-cycle responses to DNA damage in G2. Int. Rev. Cytol. 2003, 222, 99–140. [Google Scholar] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A Link between Cancer Genetics and Chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Kim, S.-K.; Cho, S.-M.; Kim, H.; Seok, H.; Kim, S.-O.; Kwon, T.K.; Chang, J.-S. The colchicine derivative CT20126 shows a novel microtubule-modulating activity with apoptosis. Exp. Mol. Med. 2013, 45, e19. [Google Scholar] [CrossRef]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef]
- Nagappan, A.; Lee, W.S.; Yun, J.W.; Lu, J.N.; Chang, S.-H.; Jeong, J.-H.; Kim, G.S.; Jung, J.-M.; Hong, S.C. Tetraarsenic hexoxide induces G2/M arrest, apoptosis, and autophagy via PI3K/Akt suppression and p38 MAPK activation in SW620 human colon cancer cells. PLoS ONE 2017, 12, e0174591. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.-L.; Gdynia, G.; Salou, M.; Radhakrishnan, P.; Duglova, K.; Heller, A.; Keim, S.; Kautz, N.; Jassowicz, A.; Elssner, C.; et al. Bcl-xL is an oncogenic driver in colorectal cancer. Cell Death Dis. 2016, 7, e2342. [Google Scholar] [CrossRef] [PubMed]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic Frameshift Mutations in the BAX Gene in Colon Cancers of the Microsatellite Mutator Phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. New Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychène, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Ikehara, N.; Semba, S.; Sakashita, M.; Aoyama, N.; Kasuga, M.; Yokozaki, H. BRAF mutation associated with dysregulation of apoptosis in human colorectal neoplasms. Int. J. Cancer 2005, 115, 943–950. [Google Scholar] [CrossRef]
- Erhardt, P.; Schremser, E.J.; Cooper, G.M. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol. Cell. Biol. 1999, 19, 5308–5315. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, W.; Jiang, X.; Liu, L.; Wei, K.; Du, H.; Wang, H.; Li, J. Anticancer effects and underlying mechanism of Colchicine on human gastric cancer cell lines in vitro and in vivo. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 930–935. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Park, Y.S.; Huh, J.W.; Lee, J.H.; Kim, H.R. shRNA against CD44 inhibits cell proliferation, invasion and migration, and promotes apoptosis of colon carcinoma cells. Oncol. Rep. 2012, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-Y.; Chen, M.; Ma, L.; Wang, X.; Chen, Y.-G.; Liu, S.-L. Role of CD44(high)/CD133(high) HCT-116 cells in the tumorigenesis of colon cancer. Oncotarget 2016, 7, 7657–7666. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Spiegelberg, D.; Glimelius, B.; Stenerlow, B.; Nestor, M. Evaluation of cancer stem cell markers CD133, CD44, CD24: Association with AKT isoforms and radiation resistance in colon cancer cells. PLoS ONE 2014, 9, e94621. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Huber, J.; Hadaschik, B.; Siegers, G.M.; Fiebig, H.H.; Schuler, J. Characterization of colon cancer cells: A functional approach characterizing CD133 as a potential stem cell marker. BMC Cancer 2012, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.; Subramaniam, V.; Rubenthiran, U.; Jothy, S. CD44 promotes resistance to apoptosis in human colon cancer cells. Exp. Mol. Pathol. 2004, 77, 18–25. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, S.; Li, Q.; Ji, Q.; Guo, P.; Liu, X. MALAT1: A long non-coding RNA highly associated with human cancers. Oncol. Lett. 2018, 16, 19–26. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Hur, K.; Toden, S.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Kusunoki, M.; Boland, C.R.; et al. Metastasis-associated long non-coding RNA drives gastric cancer development and promotes peritoneal metastasis. Carcinogenesis 2014, 35, 2731–2739. [Google Scholar] [CrossRef]
- Wu, X.S.; Wang, X.A.; Wu, W.G.; Hu, Y.P.; Li, M.L.; Ding, Q.; Weng, H.; Shu, Y.J.; Liu, T.Y.; Jiang, L.; et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 2014, 15, 806–814. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Yang, M.-H.; Hu, Z.-Y.; Xu, C.; Xie, L.-Y.; Wang, X.-Y.; Chen, S.-Y.; Li, Z.-G. MALAT1 promotes colorectal cancer cell proliferation/migration/invasion via PRKA kinase anchor protein 9. Biochim Biophys Acta 2015, 1852, 166–174. [Google Scholar] [CrossRef]
- Wu, C.; Zhu, X.; Tao, K.; Liu, W.; Ruan, T.; Wan, W.; Zhang, C.; Zhang, W. MALAT1 promotes the colorectal cancer malignancy by increasing DCP1A expression and miR203 downregulation. Mol. Carcinog. 2018, 57, 1421–1431. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Schetter, A.J.; Okayama, H.; Harris, C.C. The role of microRNAs in colorectal cancer. Cancer J. 2012, 18, 244–252. [Google Scholar] [CrossRef]
- Cekaite, L.; Eide, P.W.; Lind, G.E.; Skotheim, R.I.; Lothe, R.A. MicroRNAs as growth regulators, their function and biomarker status in colorectal cancer. Oncotarget 2016, 7, 6476–6505. [Google Scholar] [CrossRef]
- Jing, N.; Yin, L.; Sun, J.; Cao, Z.; Mao, W. Expression levels of miR-205 and miR-506 in colon cancer tissues and their relationships with clinicopathological features. Oncol. Lett. 2018, 16, 4331–4336. [Google Scholar] [CrossRef]
- Orang, A.V.; Safaralizadeh, R.; Hosseinpour Feizi, M.A.; Somi, M.H. Diagnostic and prognostic value of miR-205 in colorectal cancer. Asian Pac. J. Cancer Prev. 2014, 15, 4033–4037. [Google Scholar] [CrossRef][Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Kim, H.; Ahn, T.S.; Kim, C.-J.; Bae, S.B.; Kim, H.J.; Lee, C.-S.; Kim, T.H.; Im, J.; Lee, S.H.; Son, M.W.; et al. Oncogenic function of angiopoietin-2 in vitro and its modulation of tumor progression in colorectal carcinoma. Oncol. Lett. 2017, 14, 553–560. [Google Scholar] [CrossRef][Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nature reviews. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y.-H. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018, 32, 317–326. [Google Scholar] [CrossRef]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Arch. Int. J. Pathol. 2019, 474, 421–432. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Labib, M.B.; Philoppes, J.N.; Lamie, P.F.; Ahmed, E.R. Azole-hydrazone derivatives: Design, synthesis, in vitro biological evaluation, dual EGFR/HER2 inhibitory activity, cell cycle analysis and molecular docking study as anticancer agents. Bioorg. Chem. 2018, 76, 67–80. [Google Scholar] [CrossRef]
- Shaker, O.G.; Senousy, M.A.; Elbaz, E.M. Association of rs6983267 at 8q24, HULC rs7763881 polymorphisms and serum lncRNAs CCAT2 and HULC with colorectal cancer in Egyptian patients. Sci. Rep. 2017, 7, 16246. [Google Scholar] [CrossRef]
- Duan, W.; Du, L.; Jiang, X.; Wang, R.; Yan, S.; Xie, Y.; Yan, K.; Wang, Q.; Wang, L.; Zhang, X.; et al. Identification of a serum circulating lncRNA panel for the diagnosis and recurrence prediction of bladder cancer. Oncotarget 2016, 7, 78850–78858. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | SBET (m2/g) | Total Pore Volume a (cm3/g) | P, COL, CG-FA Content Calculation from Weight Loss (wt%) b |
|---|---|---|---|
| MSNs | 380.1 | 0.772 | 3.3 |
| MSNsP | 202.1 | 0.489 | 6.98 as P |
| MSNsPCOL | 181.8 | 0.467 | 3.60 as COL |
| MSNsPCOL/CG-FA | 89.5 | 0.352 | 33.48 as CG-FA |
| Colchicine Delivery Method | Structure | Cancer and Normal Cell Lines | Targeting Specifications |
|---|---|---|---|
| COL (free) |  | Normal cell line Cancer cell lines | High cytotoxicity (~60%) High anticancer effect at high concentration Non-targeting |
| MSNsPCOL COL-loaded silica spheres |  | Normal cell line Cancer cell lines | Less cytotoxicity (~15%) High anticancer effect Not enough cancer targeting |
| MSNsPCOL/CG-FA COL-loaded silica spheres coated with chitosan and glycine and conjugated to folic acid |  | Normal cell line Cancer cell lines | Negligible cytotoxicity (~5%) High anticancer effect High cancer-targeting Specific killing of HCT116 colon cancer (achieved 100% inhibition), PC3 prostate, and HepG2 liver cells (~80% inhibition) Cancer targeting through folate receptors Enhanced apoptosis mechanism, genetic regulation, and cancer immunotherapy effects |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AbouAitah, K.; Hassan, H.A.; Swiderska-Sroda, A.; Gohar, L.; Shaker, O.G.; Wojnarowicz, J.; Opalinska, A.; Smalc-Koziorowska, J.; Gierlotka, S.; Lojkowski, W. Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles. Cancers 2020, 12, 144. https://doi.org/10.3390/cancers12010144
AbouAitah K, Hassan HA, Swiderska-Sroda A, Gohar L, Shaker OG, Wojnarowicz J, Opalinska A, Smalc-Koziorowska J, Gierlotka S, Lojkowski W. Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles. Cancers. 2020; 12(1):144. https://doi.org/10.3390/cancers12010144
Chicago/Turabian StyleAbouAitah, Khaled, Heba A. Hassan, Anna Swiderska-Sroda, Lamiaa Gohar, Olfat G. Shaker, Jacek Wojnarowicz, Agnieszka Opalinska, Julita Smalc-Koziorowska, Stanislaw Gierlotka, and Witold Lojkowski. 2020. "Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles" Cancers 12, no. 1: 144. https://doi.org/10.3390/cancers12010144
APA StyleAbouAitah, K., Hassan, H. A., Swiderska-Sroda, A., Gohar, L., Shaker, O. G., Wojnarowicz, J., Opalinska, A., Smalc-Koziorowska, J., Gierlotka, S., & Lojkowski, W. (2020). Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles. Cancers, 12(1), 144. https://doi.org/10.3390/cancers12010144