Targeting Non-Oncogene Addiction: Focus on Thyroid Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Non-Oncogene Addiction (NOA) and Tools for Its Identification in Cancer Cells
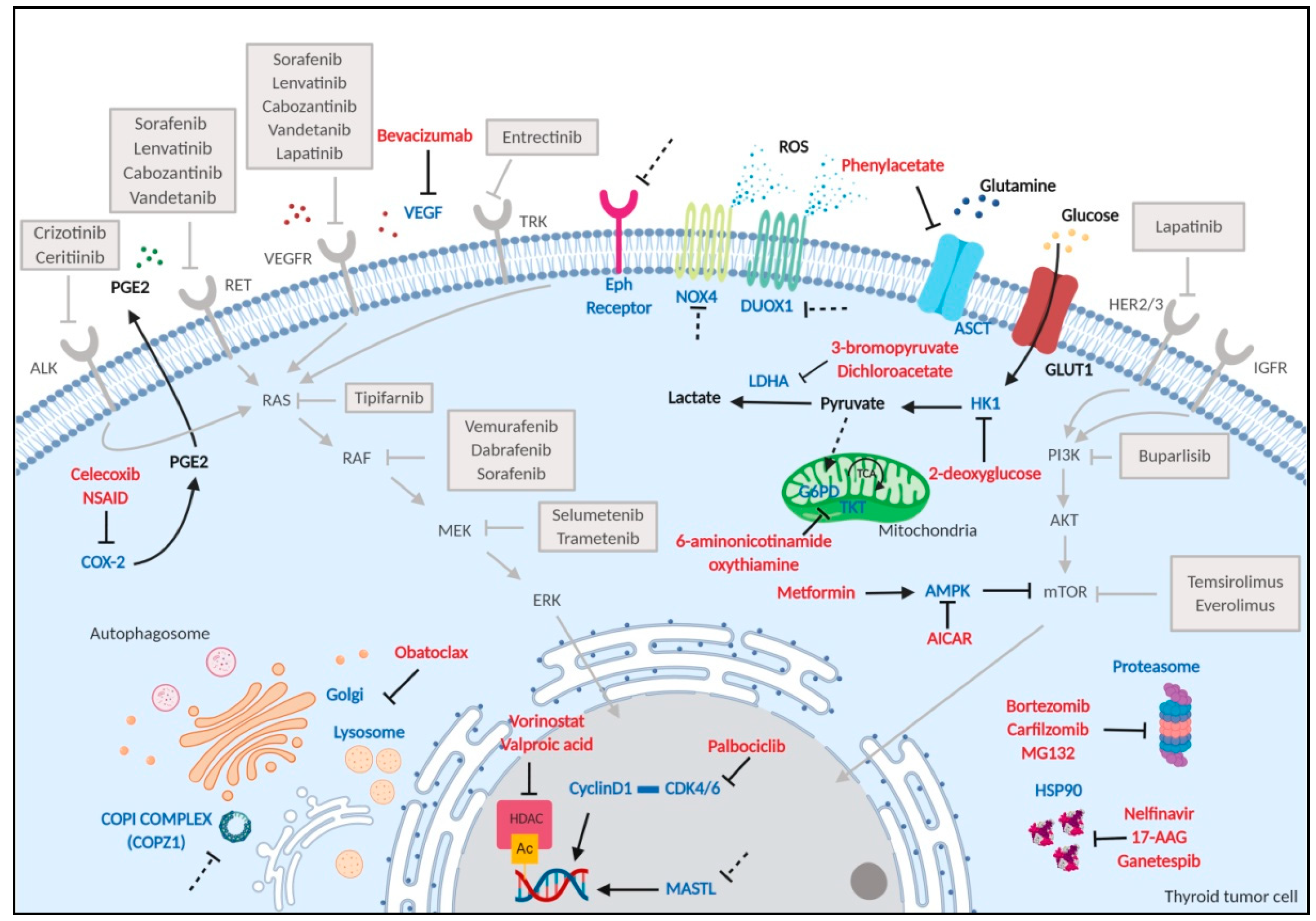
2. Thyroid Cancer
2.1. Molecular Alterations and Therapeutic Implications
2.2. Management of Thyroid Cancer Patients
3. Discovery of Vulnerabilities in Thyroid Cancer: Our Experience
3.1. Functional Screening
3.2. Validation of NOA Targets
4. Other Putative NOAs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Yoda, S.; Dagogo-Jack, I.; Hata, A.N. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol. Ther. 2019, 193, 20–30. [Google Scholar] [CrossRef]
- Von Mehren, M.; Joensuu, H. Gastrointestinal Stromal Tumors. J. Clin Oncol. 2018, 36, 136–143. [Google Scholar] [CrossRef]
- Hughes, T.P.; Kaeda, J.; Branford, S.; Rudzki, Z.; Hochhaus, A.; Hensley, M.L.; Gathmann, I.; Bolton, A.E.; van Hoomissen, I.C.; Goldman, J.M.; et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2003, 349, 1423–1432. [Google Scholar] [CrossRef]
- Valerio, L.; Pieruzzi, L.; Giani, C.; Agate, L.; Bottici, V.; Lorusso, L.; Cappagli, V.; Puleo, L.; Matrone, A.; Viola, D.; et al. Targeted Therapy in Thyroid Cancer: State of the Art. Clin Oncol. 2017, 29, 316–324. [Google Scholar] [CrossRef]
- Bronte, G.; Ulivi, P.; Verlicchi, A.; Cravero, P.; Delmonte, A.; Crino, L. Targeting RET-rearranged non-small-cell lung cancer: Future prospects. Lung Cancer 2019, 10, 27–36. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiao, Z.; Gu, W.Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.; Wang, G.; Tao, Z.F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef]
- Kennedy, R.D.; Chen, C.C.; Stuckert, P.; Archila, E.M.; Michelle, A.; Moreau, L.A.; Shimamura, A.; D’Andrea, A.D. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Investig. 2007, 117, 1440–1449. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Putcha, P.; Yu, J.; Rodriguez-Barrueco, R.; Saucedo-Cuevas, L.; Villagrasa, P.; Murga-Penas, E.; Quayle, S.N.; Yang, M.; Castro, V.; Llobet-Navas, D. HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers. Breast Cancer Res. 2015, 17, 149. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiang, Y.L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Kushchayeva, Y.; Jensen, K.; Burman, K.D.; Vasko, V. Repositioning therapy for thyroid cancer: New insights on established medications. Endocr. Relat. Cancer 2014, 21, R183–R194. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Zheng, M.; Jiang, J.; Tang, Y.L.; Liang, X.H. Oncogene and non-oncogene addiction in inflammation-associated cancers. Future Oncol. 2013, 9, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Marran, K.; Parker, J.S.; Silva, J.; Golding, M.; Schlabach, M.R.; Elledge, S.J.; Hannon, G.J.; Chang, K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 2008, 319, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Schlabach, M.R.; Luo, J.; Solimini, N.L.; Hu, G.; Xu, Q.; Li, M.Z.; Zhao, Z.; Smogorzewska, A.; Sowa, M.E.; Ang, X.L.; et al. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar] [CrossRef]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Scholl, C.; Frohling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef]
- Colombi, M.; Molle, K.D.; Benjamin, D.; Rattenbacher-Kiser, K.; Schaefer, C.; Betz, C.; Thiemeyer, A.; Regenass, U.; Hall, M.N.; Moroni, C. Genome-wide shRNA screen reveals increased mitochondrial dependence upon mTORC2 addiction. Oncogene 2011, 30, 1551–1565. [Google Scholar] [CrossRef]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef]
- Sethi, G.; Pathak, H.B.; Zhang, H.; Zhou, Y.; Einarson, M.B.; Vathipadiekal, V.; Gunewardena, S.; Birrer, M.J.; Godwin, A.K. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS ONE 2012, 7, e47086. [Google Scholar] [CrossRef] [PubMed]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.J.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140045. [Google Scholar]
- McDonald, E.R., III; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Hjaltelin, J.X.; Izarzugaza, J.M.G.; Jensen, L.J.; Russo, F.; Westergaard, D.; Brunak, S. Identification of hyper-rewired genomic stress non-oncogene addiction genes across 15 cancer types. NPJ Syst. Biol. Appl. 2019, 5, 27. [Google Scholar] [CrossRef]
- D’Alesio, C.; Punzi, S.; Cicalese, A.; Fornasari, L.; Furia, L.; Riva, L.; Carugo, A.; Curigliano, G.; Criscitiello, C.; Pruneri, G.; et al. RNAi screens identify CHD4 as an essential gene in breast cancer growth. Oncotarget 2016, 7, 80901–80915. [Google Scholar] [CrossRef]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef]
- Rudalska, R.; Dauch, D.; Longerich, T.; McJunkin, K.; Wuestefeld, T.; Kang, T.W.; Hohmeyer, A.; Pesic, M.; Leibold, J.; von Thun, A.; et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 2014, 20, 1138–1146. [Google Scholar] [CrossRef]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef]
- Fraietta, I.; Gasparri, F. The development of high-content screening (HCS) technology and its importance to drug discovery. Expert Opin. Drug Discov. 2016, 11, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Tirrò, E.; Martorana, F.; Romano, C.; Vitale, S.R.; Motta, G.; Di Gregorio, S.; Massimino, M.; Pennisi, M.S.; Stella, S.; Puma, A.; et al. Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice. Genes 2019, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Miranda, C.; Pierotti, M.A. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol. Cell. Endocrinol. 2010, 321, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- The Cancer Gene Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS Mutations Cooperate to Drive Thyroid Tumorigenesis through ATF4 and c-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef]
- Pishkari, S.; Paryan, M.; Hashemi, M.; Baldini, E.; Mohammadi-Yeganeh, S. The role of microRNAs in different types of thyroid carcinoma: A comprehensive analysis to find new miRNA supplementary therapies. J. Endocrinol. Investig. 2018, 41, 269–283. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Santisteban, P. miRNA-Directed Regulation of the Main Signaling Pathways in Thyroid Cancer. Front. Endocrinol. 2019, 10, 430. [Google Scholar] [CrossRef]
- Sedaghati, M.; Kebebew, E. Long noncoding RNAs in thyroid cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 275–281. [Google Scholar] [CrossRef]
- Mahmoudian-Sani, M.R.; Jalali, A.; Jamshidi, M.; Moridi, H.; Alghasi, A.; Shojaeian, A.; Mobini, G.R. Long Non-Coding RNAs in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, and Therapy. Oncol. Res. Treat. 2019, 42, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Degl’Innocenti, D.; Romeo, P.; Tarantino, E.; Sensi, M.; Cassinelli, G.; Catalano, V.; Lanzi, C.; Perrone, F.; Pilotti, S.; Seregni, E.; et al. DUSP6/MKP3 is overexpressed in papillary and poorly differentiated thyroid carcinoma and contributes to neoplastic properties of thyroid cancer cells. Endocr. Relat. Cancer 2013, 20, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Prasad, M.; Lemon, W.J.; Hampel, H.; Wright, F.A.; Kornacker, K.; LiVolsi, V.; Frankel, W.; Kloos, R.T.; Eng, C.; et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 15044–15049. [Google Scholar] [CrossRef] [PubMed]
- Anania, M.C.; Miranda, C.; Vizioli, M.G.; Mazzoni, M.; Cleris, L.; Pagliardini, S.; Manenti, G.; Borrello, M.G.; Pierotti, M.A.; Greco, A. S100A11 Overexpression Contributes to the Malignant Phenotype of Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E1591–E1600. [Google Scholar] [CrossRef] [PubMed]
- Salerno, P.; Garcia-Rostan, G.; Piccinin, S.; Bencivenga, T.C.; Di, M.G.; Doglioni, C.; Basolo, F.; Maestro, R.; Fusco, A.; Santoro, M.; et al. TWIST1 plays a pleiotropic role in determining the anaplastic thyroid cancer phenotype. J. Clin. Endocrinol. Metab. 2011, 96, E772–E781. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xing, M. Gene methylation in thyroid tumorigenesis. Endocrinology 2007, 148, 948–953. [Google Scholar] [CrossRef]
- Zhang, K.; Li, C.; Liu, J.; Tang, X.; Li, Z. DNA methylation alterations as therapeutic prospects in thyroid cancer. J. Endocrinol. Investig. 2019, 42, 363–370. [Google Scholar] [CrossRef]
- Anania, M.C.; Sensi, M.; Radaelli, E.; Miranda, C.; Vizioli, M.G.; Pagliardini, S.; Favini, E.; Cleris, L.; Supino, R.; Formelli, F.; et al. TIMP3 regulates migration, invasion and in vivo tumorigenicity of thyroid tumor cells. Oncogene 2011, 30, 3011–3023. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Sensi, M.; Miranda, C.; Cleris, L.; Formelli, F.; Anania, M.C.; Pierotti, M.A.; Greco, A. IGFBP7: An oncosuppressor gene in thyroid carcinogenesis. Oncogene 2010, 29, 3835–3844. [Google Scholar] [CrossRef]
- Alvarez-Nuñez, F.; Bussaglia, E.; Mauricio, D.; Ybarra, J.; Vilar, M.; Lerma, E.; Leiva, A.D.; Matias-Guiu, X. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 2006, 16, 17–23. [Google Scholar] [CrossRef]
- Ferrario, C.; Lavagni, P.; Gariboldi, M.; Miranda, C.; Losa, M.; Cleris, L.; Formelli, F.; Pilotti, S.; Pierotti, M.A.; Greco, A. Metallothionein 1G acts as an oncosupressor in papillary thyroid carcinoma. Lab. Investig. 2008, 88, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Wang, R.; Zou, K.; Zou, L. Genetic alterations in anaplastic thyroid carcinoma and targeted therapies. Exp. Ther. Med. 2019, 18, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Eustatia-Rutten, C.F.; Corssmit, E.P.; Biermasz, N.R.; Pereira, A.M.; Romijn, J.A.; Smit, J.W. Survival and death causes in differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dohan, O.; De la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide Symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar] [CrossRef]
- Anania, M.C.; Gasparri, F.; Cetti, E.; Fraietta, I.; Todoerti, K.; Miranda, C.; Mazzoni, M.; Re, C.; Colombo, R.; Ukmar, G.; et al. Identification of thyroid tumor cell vulnerabilities through a siRNA-based functional screening. Oncotarget 2015, 6, 34629–34648. [Google Scholar] [CrossRef]
- Cantisani, M.C.; Parascandolo, A.; Perala, M.; Allocca, C.; Fey, V.; Sahlberg, N.; Merolla, F.; Basolo, F.; Laukkanen, M.O.; Kallioniemi, O.P.; et al. A loss-of-function genetic screening identifies novel mediators of thyroid cancer cell viability. Oncotarget 2016, 7, 28510. [Google Scholar] [CrossRef][Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Seybt, T.P.; Ramalingam, P.; Huang, J.; Looney, S.W.; Reid, M.D. Cyclin D1 expression in benign and differentiated malignant tumors of the thyroid gland: Diagnostic and biologic implications. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 124–130. [Google Scholar] [CrossRef]
- Lee, J.J.; Au, A.Y.; Foukakis, T.; Barbaro, M.; Kiss, N.; Clifton-Bligh, R.; Staaf, J.; Borg, A.; Delbridge, L.; Robinson, B.G.; et al. Array-CGH identifies cyclin D1 and UBCH10 amplicons in anaplastic thyroid carcinoma. Endocr. Relat. Cancer 2008, 15, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Khoo, M.L.; Ezzat, S.; Freeman, J.L.; Asa, S.L. Cyclin D1 protein expression predicts metastatic behavior in thyroid papillary microcarcinomas but is not associated with gene amplification. J. Clin. Endocrinol. Metab. 2002, 87, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Hryhorowicz, S.; Ziemnicka, K.; Kaczmarek-Rys, M.; Hoppe-Golebiewska, J.; Plawski, A.; Skrzypczak-Zielinska, M.; Szkudlarek, M.; Golab, M.; Budny, B.; Ruchala, M.; et al. CCND1 gene polymorphic variants in patients with differentiated thyroid carcinoma. Oncol. Lett. 2015, 9, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Kim, Y.; Jeong, Y.M.; Bae, J.S.; Jung, C.K. CCND1 Splice Variant as A Novel Diagnostic and Predictive Biomarker for Thyroid Cancer. Cancers 2018, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Sun, F. Identification of key genes of papillary thyroid cancer using integrated bioinformatics analysis. J. Endocrinol. Investig. 2018, 41, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Hong, S.; Yu, S.; Huang, Y.; Chen, S.; Liu, Y.; Zhang, Q.; Li, Y.; Xiao, H. MiR-195 Inhibits Tumor Growth and Metastasis in Papillary Thyroid Carcinoma Cell Lines by Targeting CCND1 and FGF2. Int. J. Endocrinol. 2017, 2017, 6180425. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Fu, Q.; Wang, Y.; Sui, G. Long non-coding RNA NR2F1-AS1 promoted proliferation and migration yet suppressed apoptosis of thyroid cancer cells through regulating miRNA-338-3p/CCND1 axis. J. Cell Mol. Med. 2019, 23, 5907–5919. [Google Scholar] [CrossRef]
- Sun, J.; Shi, R.; Zhao, S.; Li, X.; Lu, S.; Bu, H.; Ma, X.; Su, C. E2F8, a direct target of miR-144, promotes papillary thyroid cancer progression via regulating cell cycle. J. Exp. Clin. Cancer Res. 2017, 36, 40. [Google Scholar] [CrossRef]
- Wong, K.; Di, C.F.; Ranieri, M.; De, M.D.; Di, C.A. PI3K/mTOR inhibition potentiates and extends palbociclib activity in anaplastic thyroid cancer. Endocr. Relat. Cancer 2019, 26, 425–436. [Google Scholar] [CrossRef]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The efficacy of HRAS and CDK4/6 inhibitors in anaplastic thyroid cancer cell lines. J. Endocrinol. Investig. 2019, 42, 527–540. [Google Scholar] [CrossRef]
- Antonello, Z.A.; Hsu, N.; Bhasin, M.; Roti, G.; Joshi, M.; Van, H.P.; Ye, E.; Lo, A.S.; Karumanchi, S.A.; Bryke, C.R.; et al. Vemurafenib-resistance via de novo RBM genes mutations and chromosome 5 aberrations is overcome by combined therapy with palbociclib in thyroid carcinoma with BRAF(V600E). Oncotarget 2017, 8, 84743–84760. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. Entry into mitosis: A solution to the decades-long enigma of MPF. Chromosoma 2015, 124, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Peng, A.; Yamamoto, T.M.; Goldberg, M.L.; Maller, J.L. A novel role for greatwall kinase in recovery from DNA damage. Cell Cycle 2010, 9, 4364–4369. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.Y.; Ma, H.T.; Lee, H.J.; Poon, R.Y. MASTL (Greatwall) regulates DNA damage responses by coordinating mitotic entry after checkpoint recovery and APC/C activation. Sci. Rep. 2016, 6, 22230. [Google Scholar] [CrossRef] [PubMed]
- Marzec, K.; Burgess, A. The Oncogenic Functions of MASTL Kinase. Front. Cell Dev. Biol. 2018, 6, 162. [Google Scholar] [CrossRef]
- Wang, L.; Luong, V.Q.; Giannini, P.J.; Peng, A. Mastl kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget 2014, 5, 11479–11489. [Google Scholar] [CrossRef]
- Vera, J.; Lartigue, L.; Vigneron, S.; Gadea, G.; Gire, V.; Del Rio, M.; Soubeyran, I.; Chibon, F.; Lorca, T.; Castro, A. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. eLife 2015, 4, e10115. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.J.; Yang, J.H.; Wang, Y.; Hua, Z.J.; Liu, D.; Chen, Y.Q.; Zhang, H.M.; Zhang, R.; Zhao, J.S.; et al. Inflammatory cytokine-induced expression of MASTL is involved in hepatocarcinogenesis by regulating cell cycle progression. Oncol. Lett. 2019, 17, 3163–3172. [Google Scholar] [CrossRef]
- Alvarez-Fernandez, M.; Sanz-Flores, M.; Sanz-Castillo, B.; Salazar-Roa, M.; Partida, D.; Zapatero-Solana, E.; Ali, H.R.; Manchado, E.; Lowe, S.; VanArsdale, T.; et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2018, 25, 828–840. [Google Scholar] [CrossRef]
- Rogers, S.; McCloy, R.A.; Parker, B.L.; Gallego-Ortega, D.; Law, A.M.K.; Chin, V.T.; Conway, J.R.W.; Fey, D.; Millar, E.K.A.; O’Toole, S.; et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 2018, 37, 4518–4533. [Google Scholar] [CrossRef]
- Cetti, E.; Di Marco, T.; Mauro, G.; Mazzoni, M.; Lecis, D.; Minna, E.; Gioiosa, L.; Brich, S.; Pagliardini, S.; Borrello, M.G.; et al. Mitosis perturbation by MASTL depletion impairs the viability of thyroid tumor cells. Cancer Lett. 2019, 442, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.A.; Kang, K.S.; Di Cristofano, A. The PLK1 inhibitor GSK461364A is effective in poorly differentiated and anaplastic thyroid carcinoma cells, independent of the nature of their driver mutations. Thyroid 2013, 23, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Baldini, E.; Tuccilli, C.; Prinzi, N.; Sorrenti, S.; Antonelli, A.; Gnessi, L.; Morrone, S.; Moretti, C.; Bononi, M.; Arlot-Bonnemains, Y.; et al. Effects of selective inhibitors of Aurora kinases on anaplastic thyroid carcinoma cell lines. Endocr. Relat. Cancer 2014, 21, 797–811. [Google Scholar] [CrossRef]
- Uppada, S.B.; Gowrikumar, S.; Ahmad, R.; Kumar, B.; Szeglin, B.; Chen, X.; Smith, J.J.; Batra, S.K.; Singh, A.B.; Dhawan, P. MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/beta-catenin signaling. Mol. Cancer 2018, 17, 111. [Google Scholar] [CrossRef]
- Nagel, R.; Stigter-van Walsum, M.; Buijze, M.; van den Berg, J.; van der Meulen, I.H.; Hodzic, J.; Piersma, S.R.; Pham, T.V.; Jiménez, C.R.; van Beusechem, V.W.; et al. Genome-wide siRNA screen identifies the radiosensitizing effect of downregulation of MASTL and FOXM1 in NSCLC. Mol. Cancer Ther. 2015, 14, 1434–1444. [Google Scholar] [CrossRef]
- Yoon, Y.N.; Choe, M.H.; Jung, K.Y.; Hwang, S.G.; Oh, J.S.; Kim, J.S. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer 2018, 18, 716. [Google Scholar] [CrossRef]
- Alvarez-Fernandez, M.; Sanchez-Martinez, R.; Sanz-Castillo, B.; Gan, P.P.; Sanz-Flores, M.; Trakala, M.; Ruiz-Torres, M.; Lorca, T.; Castro, A.; Malumbres, M. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc. Natl. Acad. Sci. USA 2013, 110, 17374–17379. [Google Scholar] [CrossRef]
- Ocasio, C.A.; Rajasekaran, M.B.; Walker, S.; Le, G.D.; Spencer, J.; Pearl, F.M.; Ward, S.E.; Savic, V.; Pearl, L.H.; Hochegger, H.; et al. A first generation inhibitor of human Greatwall kinase, enabled by structural and functional characterisation of a minimal kinase domain construct. Oncotarget 2016, 7, 71182–71197. [Google Scholar] [CrossRef]
- Ammarah, U.; Kumar, A.; Pal, R.; Bal, N.C.; Misra, G. Identification of new inhibitors against human Great wall kinase using in silico approaches. Sci. Rep. 2018, 8, 4894. [Google Scholar] [CrossRef]
- Beck, R.; Rawet, M.; Wieland, F.T.; Cassel, D. The COPI system: Molecular mechanisms and function. FEBS Lett. 2009, 583, 2701–2709. [Google Scholar] [CrossRef] [PubMed]
- Razi, M.; Chan, E.Y.; Tooze, S.A. Early endosomes and endosomal coatomer are required for autophagy. J. Cell Biol. 2009, 185, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 19036–19041. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Sztalryd, C.; Southall, N.; Bell, M.; Jackle, H.; Auld, D.S.; Oliver, B. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008, 6, e292. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Baig, M.; Levina, E.; Hurteau, G.; Lim, C.U.; Broude, E.; Nikiforov, M.; Harkins, T.T.; Carmack, C.S.; Ding, Y.; et al. Tumor-specific silencing of COPZ2 gene encoding coatomer protein complex subunit zeta 2 renders tumor cells dependent on its paralogous gene COPZ1. Proc. Natl. Acad. Sci. USA 2011, 108, 12449–12454. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Roninson, I.B. A subunit of coatomer protein complex offers a novel tumor-specific target through a surprising mechanism. Autophagy 2011, 7, 1551–1552. [Google Scholar] [CrossRef]
- Anania, M.C.; Cetti, E.; Lecis, D.; Todoerti, K.; Gulino, A.; Mauro, G.; Di Marco, T.; Cleris, L.; Pagliardini, S.; Manenti, G.; et al. Targeting COPZ1 non-oncogene addiction counteracts the viability of thyroid tumor cells. Cancer Lett. 2017, 410, 201–211. [Google Scholar] [CrossRef]
- Sudo, H.; Tsuji, A.B.; Sugyo, A.; Kohda, M.; Sogawa, C.; Yoshida, C.; Harada, Y.N.; Hino, O.; Saga, T. Knockdown of COPA, identified by loss-of-function screen, induces apoptosis and suppresses tumor growth in mesothelioma mouse model. Genomics 2010, 95, 210–216. [Google Scholar] [CrossRef]
- Oliver, D.; Ji, H.; Liu, P.; Gasparian, A.; Gardiner, E.; Lee, S.; Zenteno, A.; Perinskaya, L.O.; Chen, M.; Buckhaults, P.; et al. Identification of novel cancer therapeutic targets using a designed and pooled shRNA library screen. Sci. Rep. 2017, 7, 43023. [Google Scholar] [CrossRef]
- Kim, H.S.; Mendiratta, S.; Kim, J.; Pecot, C.V.; Larsen, J.E.; Zubovych, I.; Seo, B.Y.; Kim, J.; Eskiocak, B.; Chung, H.; et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 2013, 155, 552–566. [Google Scholar] [CrossRef]
- Pu, X.; Wang, J.; Li, W.; Fan, W.; Wang, L.; Mao, Y.; Yang, S.; Liu, S.; Xu, J.; Lv, Z.; et al. COPB2 promotes cell proliferation and tumorigenesis through up-regulating YAP1 expression in lung adenocarcinoma cells. Biomed. Pharm. 2018, 103, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.S.; Liu, C.H.; Liu, Z.; Zhu, C.L.; Huang, Q. Downregulation of COPB2 by RNAi inhibits growth of human cholangiocellular carcinoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 985–992. [Google Scholar] [PubMed]
- An, C.; Li, H.; Zhang, X.; Wang, J.; Qiang, Y.; Ye, X.; Li, Q.; Guan, Q.; Zhou, Y. Silencing of COPB2 inhibits the proliferation of gastric cancer cells and induces apoptosis via suppression of the RTK signaling pathway. Int. J. Oncol. 2019, 54, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Okamura, M.; Hirosawa, A.; Tamaki, N.; Akatsuka, A.; Wu, K.M.; Choi, H.W.; Yoshimatsu, K.; Shiina, I.; Yamori, T.; et al. M-COPA, a Golgi Disruptor, Inhibits Cell Surface Expression of MET Protein and Exhibits Antitumor Activity against MET-Addicted Gastric Cancers. Cancer Res. 2016, 76, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Okamura, M.; Katayama, R.; Fang, S.; Tsutsui, S.; Akatsuka, A.; Shan, M.; Choi, H.W.; Fujita, N.; Yoshimatsu, K.; et al. Targeting the Golgi apparatus to overcome acquired resistance of non-small cell lung cancer cells to EGFR tyrosine kinase inhibitors. Oncotarget 2018, 9, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef] [PubMed]
- Szanto, I.; Pusztaszeri, M.; Mavromati, M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants 2019, 8, 126. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar]
- Liu, C.L.; Hsu, Y.C.; Lee, J.J.; Chen, M.J.; Lin, C.H.; Huang, S.Y.; Cheng, S.P. Targeting the pentose phosphate pathway increases reactive oxygen species and induces apoptosis in thyroid cancer cells. Mol. Cell. Endocrinol. 2019, 499, 110595. [Google Scholar] [CrossRef]
- Spartalis, E.; Athanasiadis, D.I.; Chrysikos, D.; Spartalis, M.; Boutzios, G.; Schizas, D.; Garmpis, N.; Damaskos, C.; Paschou, S.A.; Ioannidis, A.; et al. Histone Deacetylase Inhibitors and Anaplastic Thyroid Carcinoma. Anticancer Res. 2019, 39, 1119–1127. [Google Scholar] [CrossRef]
- Sherman, S.I. Targeted therapies for thyroid tumors. Mod. Pathol. 2011, 24 (Suppl. 2), S44–S52. [Google Scholar] [CrossRef]
- Zhang, L.; Boufraqech, M.; Lake, R.; Kebebew, E. Carfilzomib potentiates CUDC-101-induced apoptosis in anaplastic thyroid cancer. Oncotarget 2016, 7, 16517–16528. [Google Scholar] [CrossRef] [PubMed]
- Woan, K.V.; Sahakian, E.; Sotomayor, E.M.; Seto, E.; Villagra, A. Modulation of antigen-presenting cells by HDAC inhibitors: Implications in autoimmunity and cancer. Immunol. Cell Biol. 2012, 90, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Altmann, A.; Markert, A.; Askoxylakis, V.; Schoning, T.; Jesenofsky, R.; Eisenhut, M.; Haberkorn, U. Antitumor effects of proteasome inhibition in anaplastic thyroid carcinoma. J. Nucl. Med. 2012, 53, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Sui, F.; Ma, J.; Li, X.; Ren, X.; Shao, Y.; Liu, J.; Guan, H.; Shi, B.; Hou, P. Proteasome inhibitor MG132 induces thyroid cancer cell apoptosis by modulating the activity of transcription factor FOXO3a. Endocrine 2017, 56, 98–108. [Google Scholar] [CrossRef]
- Mehta, A.; Zhang, L.; Boufraqech, M.; Zhang, Y.; Patel, D.; Shen, M.; Kebebew, E. Carfilzomib is an effective anticancer agent in anaplastic thyroid cancer. Endocr. Relat. Cancer 2015, 22, 319–329. [Google Scholar] [CrossRef]
- Shim, J.S.; Rao, R.; Beebe, K.; Neckers, L.; Han, I.; Nahta, R.; Liu, J.O. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J. Natl. Cancer Inst. 2012, 104, 1576–1590. [Google Scholar] [CrossRef]
- Driessen, C.; Muller, R.; Novak, U.; Cantoni, N.; Betticher, D.; Mach, N.; Rufer, A.; Mey, U.; Samaras, P.; Ribi, K.; et al. Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma. Blood 2018, 132, 2097–2100. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Coucke, C.; Laukens, D.; Devisscher, L.; Paridaens, A.; Bogaerts, E.; Vandierendonck, A.; Raevens, S.; Verhelst, X.; Van, S.C.; et al. Next-generation proteasome inhibitor oprozomib synergizes with modulators of the unfolded protein response to suppress hepatocellular carcinoma. Oncotarget 2016, 7, 34988–35000. [Google Scholar] [CrossRef]
- Abt, D.; Besse, A.; Sedlarikova, L.; Kraus, M.; Bader, J.; Silzle, T.; Vodinska, M.; Slaby, O.; Schmid, H.P.; Engeler, D.S.; et al. Improving the efficacy of proteasome inhibitors in the treatment of renal cell carcinoma by combination with the human immunodeficiency virus (HIV)-protease inhibitors lopinavir or nelfinavir. BJU Int. 2018, 121, 600–609. [Google Scholar] [CrossRef]
- Jensen, K.; Bikas, A.; Patel, A.; Kushchayeva, Y.; Costello, J.; McDaniel, D.; Burman, K.; Vasko, V. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr. Relat. Cancer 2017, 24, 147–156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Braga-Basaria, M.; Hardy, E.; Gottfried, R.; Burman, K.D.; Saji, M.; Ringel, M.D. 17-Allylamino-17-demethoxygeldanamycin activity against thyroid cancer cell lines correlates with heat shock protein 90 levels. J. Clin. Endocrinol. Metab. 2004, 89, 2982–2988. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Lin, J.D.; Hsueh, C.; Chou, T.C.; Yeh, C.N.; Chen, M.H.; Wong, R.J. Efficacy of an HSP90 inhibitor, ganetespib, in preclinical thyroid cancer models. Oncotarget 2017, 8, 41294–41304. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.S.; Zhang, T.T.; Xue, D.X.; Wu, W.L.; Wang, Y.L.; Wang, Y.; Ji, Q.H.; Zhu, Y.X.; Qu, N.; Shi, R.L. Metabolic reprogramming and its clinical application in thyroid cancer. Oncol. Lett. 2019, 18, 1579–1584. [Google Scholar] [CrossRef]
- Coelho, R.G.; Fortunato, R.S.; Carvalho, D.P. Metabolic Reprogramming in Thyroid Carcinoma. Front. Oncol. 2018, 8, 82. [Google Scholar] [CrossRef]
- Champa, D.; Russo, M.A.; Liao, X.H.; Refetoff, S.; Ghossein, R.A.; Di Cristofano, A. Obatoclax overcomes resistance to cell death in aggressive thyroid carcinomas by countering Bcl2a1 and Mcl1 overexpression. Endocr. Relat. Cancer 2014, 21, 755–767. [Google Scholar] [CrossRef]
- Mazzoni, M.; Mauro, G.; Erreni, M.; Romeo, P.; Minna, E.; Vizioli, M.G.; Belgiovine, C.; Rizzetti, M.G.; Pagliardini, S.; Avigni, R.; et al. Senescent thyrocytes and thyroid tumor cells induce M2-like macrophage polarization of human monocytes via a PGE2-dependent mechanism. J. Exp. Clin. Cancer Res. 2019, 38, 208. [Google Scholar] [CrossRef]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef]
- Mrozek, E.; Kloos, R.T.; Ringel, M.D.; Kresty, L.; Snider, P.; Arbogast, D.; Kies, M.; Munden, R.; Busaidy, N.; Klein, M.J.; et al. Phase II study of celecoxib in metastatic differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 2201–2204. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anania, M.C.; Di Marco, T.; Mazzoni, M.; Greco, A. Targeting Non-Oncogene Addiction: Focus on Thyroid Cancer. Cancers 2020, 12, 129. https://doi.org/10.3390/cancers12010129
Anania MC, Di Marco T, Mazzoni M, Greco A. Targeting Non-Oncogene Addiction: Focus on Thyroid Cancer. Cancers. 2020; 12(1):129. https://doi.org/10.3390/cancers12010129
Chicago/Turabian StyleAnania, Maria Chiara, Tiziana Di Marco, Mara Mazzoni, and Angela Greco. 2020. "Targeting Non-Oncogene Addiction: Focus on Thyroid Cancer" Cancers 12, no. 1: 129. https://doi.org/10.3390/cancers12010129
APA StyleAnania, M. C., Di Marco, T., Mazzoni, M., & Greco, A. (2020). Targeting Non-Oncogene Addiction: Focus on Thyroid Cancer. Cancers, 12(1), 129. https://doi.org/10.3390/cancers12010129