Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Anaplerosis and Cataplerosis
3. The Warburg Effect in Normal Cells
4. Cancer Metabolism
5. Role of the Transcription Factors STAT and HIF in the Deregulation of Energy Metabolism in Cancer Cells
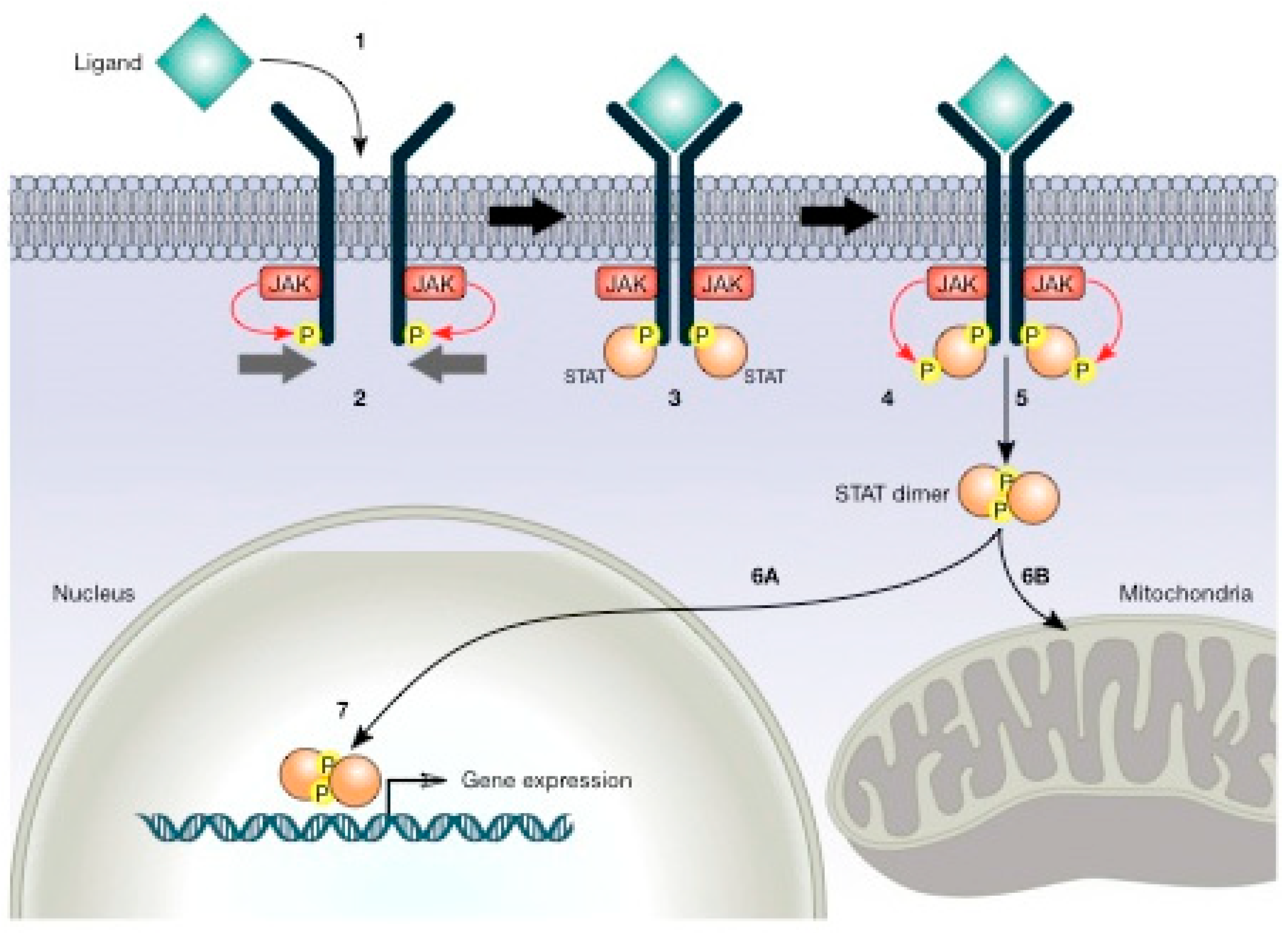
5.1. Role of STAT Proteins in Metabolism
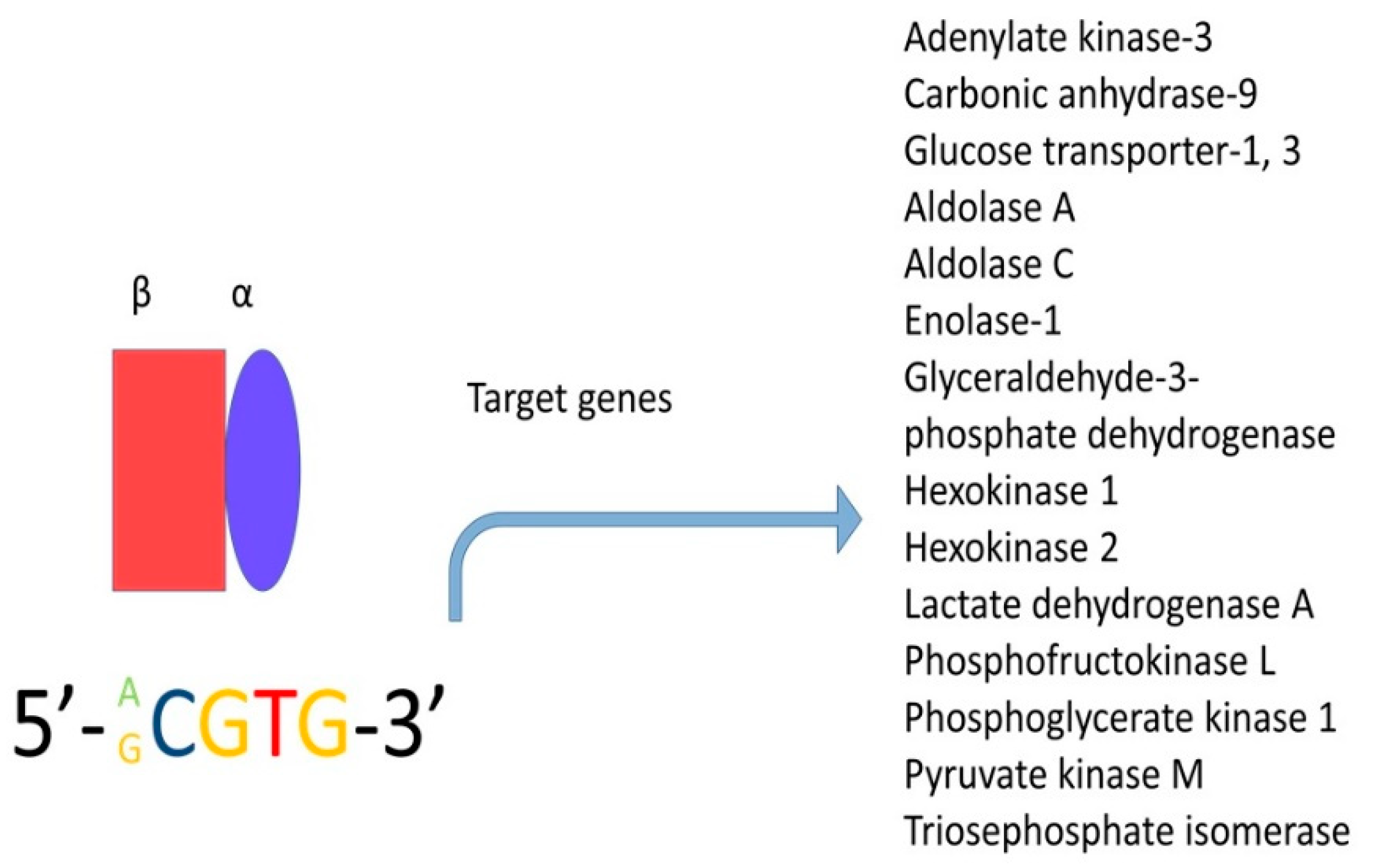
5.2. STAT3
5.3. STAT5
5.4. The Importance of Oxygen and HIF-1α in the Regulation of Metabolism
6. Conclusions
Funding
Conflicts of Interest
References
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Brown, R.W.; Mody, D.R.; Fernandez, L.; Laucirica, R. GLUT1 expression in human breast carcinoma: Correlation with known prognostic markers. Anticancer Res. 1995, 15, 2895–2898. [Google Scholar] [PubMed]
- Haber, R.S.; Weiser, K.R.; Pritsker, A.; Reder, I.; Burstein, D.E. GLUT1 glucose transporter expression in benign and malignant thyroid nodules. Thyroid 1997, 7, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Seino, Y.; Fukumoto, H.; Koh, G.; Yano, H.; Inagaki, N.; Yamada, Y.; Inoue, K.; Manabe, T.; Imura, H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem. Biophys. Res. Commun. 1990, 170, 223–230. [Google Scholar] [CrossRef]
- Kurata, T.; Oguri, T.; Isobe, T.; Ishioka, S.; Yamakido, M. Differential expression of facilitative glucose transporter (GLUT) genes in primary lung cancers and their liver metastases. Jpn. J. Cancer Res. 1999, 90, 1238–1243. [Google Scholar] [CrossRef]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef]
- Nagamatsu, S.; Sawa, H.; Wakizaka, A.; Hoshino, T. Expression of facilitative glucose transporter isoforms in human brain tumors. J. Neurochem. 1993, 61, 2048–2053. [Google Scholar] [CrossRef]
- Ito, T.; Noguchi, Y.; Satoh, S.; Hayashi, H.; Inayama, Y.; Kitamura, H. Expression of facilitative glucose transporter isoforms in lung carcinomas: Its relation to histologic type, differentiation grade, and tumor stage. Mod. Pathol. 1998, 11, 437–443. [Google Scholar]
- Noguchi, Y.; Marat, D.; Saito, A.; Yoshikawa, T.; Doi, C.; Fukuzawa, K.; Tsuburaya, A.; Satoh, S.; Ito, T. Expression of facilitative glucose transporters in gastric tumors. Hepatogastroenterology 1999, 46, 2683–2689. [Google Scholar]
- Armoni, M.; Quon, M.J.; Maor, G.; Avigad, S.; Shapiro, D.N.; Harel, C.; Esposito, D.; Goshen, Y.; Yaniv, I.; Karnieli, E. PAX3/forkhead homolog in rhabdomyosarcoma oncoprotein activates glucose transporter 4 gene expression in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 5312–5324. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, G.; Hong, L.; Zhou, L.; Hu, M.; Li, B.; Huang, J.; Xia, L.; Li, C. How does hypoxia inducible factor-1α participate in enhancing the glycolysis activity in cervical cancer? Ann. Diagn. Pathol. 2013, 17, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Das, M.R.; Bag, A.K.; Saha, S.; Ghosh, A.; Dey, S.K.; Das, P.; Mandal, C.; Ray, S.; Chakrabarti, S.; Ray, M.; et al. Molecular association of glucose-6-phosphate isomerase and pyruvate kinase M2 with glyceraldehyde-3-phosphate dehydrogenase in cancer cells. BMC Cancer 2016, 16, 152. [Google Scholar] [CrossRef] [PubMed]
- Haga, A.; Niinaka, Y.; Raz, A. Phosphohexose isomerase/autocrine motility factor/neuroleukin/maturation factor is a multifunctional phosphoprotein. Biochim. Biophys. Acta 2000, 1480, 235–244. [Google Scholar] [CrossRef]
- Somarowthu, S.; Brodkin, H.R.; D’Aquino, J.A.; Ringe, D.; Ondrechen, M.J.; Beuning, P.J. A tale of two isomerases: Compact versus extended active sites in ketosteroid isomerase and phosphoglucose isomerase. Biochemistry 2011, 50, 9283–9295. [Google Scholar] [CrossRef] [PubMed]
- Niinaka, Y.; Paku, S.; Haga, A.; Watanabe, H.; Raz, A. Expression and secretion of neuroleukin/phosphohexose isomerase/maturation factor as autocrine motility factor by tumor cells. Cancer Res. 1998, 58, 2667–2674. [Google Scholar]
- Nabi, I.R.; Watanabe, H.; Raz, A. Autocrine motility factor and its receptor: Role in cell locomotion and metastasis. Cancer Metastasis Rev. 1992, 11, 5–20. [Google Scholar] [CrossRef]
- Funasaka, T.; Yanagawa, T.; Hogan, V.; Raz, A. Regulation of phosphoglucose isomerase/autocrine motility factor expression by hypoxia. FASEB J. 2005, 19, 1422–1430. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Wang, G.; Xu, Z.; Wang, C.; Yao, F.; Li, J.; Chen, C.; Sun, S. Differential phosphofructokinase-1 isoenzyme patterns associated with glycolytic efficiency in human breast cancer and paracancer tissues. Oncol. Lett. 2013, 6, 1701–1706. [Google Scholar] [CrossRef]
- Sun, C.M.; Xiong, D.B.; Yan, Y.; Geng, J.; Liu, M.; Yao, X.D. Genetic alteration in phosphofructokinase family promotes growth of muscle-invasive bladder cancer. Int. J. Biol. Markers 2016, 31, e286–e293. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Guan, Z.; Hao, L.; Song, Y.; Wang, L.; Gong, L.; Liu, L.; Qi, X.; Hou, Z.; Shao, S. Fructose-bisphosphate aldolase a is a potential metastasis-associated marker of lung squamous cell carcinoma and promotes lung cell tumorigenesis and migration. PLoS ONE 2014, 9, e85804. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.H.; Roehrl, M.H.; Wang, J.Y. Glycoproteomic Analysis of Human Lung Adenocarcinomas Using Glycoarrays and Tandem Mass Spectrometry: Differential Expression and Glycosylation Patterns of Vimentin and Fetuin A Isoforms. Protein J. 2009, 28, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Poschmann, G.; Sitek, B.; Sipos, B.; Ulrich, A.; Wiese, S.; Stephan, C.; Warscheid, B.; Klöppel, G.; Vander Borght, A.; Ramaekers, F.C.; et al. Identification of Proteomic Differences between Squamous Cell Carcinoma of the Lung and Bronchial Epithelium. Mol. Cell. Proteom. 2009, 8, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Chaerkady, R.; Harsha, H.C.; Nalli, A.; Gucek, M.; Vivekanandan, P.; Akhtar, J.; Cole, R.N.; Simmers, J.; Schulick, R.D.; Singh, S.; et al. A quantitative proteomic approach for identification of potential biomarkers in hepatocellular carcinoma. J. Proteome Res. 2008, 7, 4289–4298. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Uemura, M.; Munakata, K.; Takahashi, H.; Haraguchi, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Ikenaga, M.; Murata, K.; et al. Fructose-bisphosphate aldolase A is a key regulator of hypoxic adaptation in colorectal cancer cells and involved in treatment resistance and poor prognosis. Int. J. Oncol. 2017, 50, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Huang, Z.; Tian, Y.; Lin, B.; He, R.; Wang, H.; Ouyang, P.; Chen, H.; Wu, L. Clinical significance and prognostic value of Triosephosphate isomerase expression in gastric cancer. Medicine 2017, 96, e6865. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Huang, C.-C.; Thomas, D.G.; Shedden, K.A.; Taylor, J.M.G.; Kardia, S.L.R.; Misek, D.E.; Giordano, T.J.; Iannettoni, M.D.; et al. Proteomic analysis of lung adenocarcinoma: Identification of a highly expressed set of proteins in tumors. Clin. Cancer Res. 2002, 8, 2298–2305. [Google Scholar]
- Montgaomerie, J.Z.; Garcy, R.W.; Holshuh, H.J.; Keyser, A.J.; Bennett, C.J.; Schick, D.G. The 28K protein in urinary bladder, squamous metaplasia and urine is triosephosphate isomerase. Clin. Biochem. 1997, 8, 613–618. [Google Scholar] [CrossRef]
- Tamesa, M.S.; Kuramitsu, Y.; Fujimoto, M.; Maeda, N.; Nagashima, Y.; Tanaka, T.; Yamamoto, S.; Oka, M.; Nakamura, K. Detection of autoantibodies against cyclophilin A and triosephosphate isomerase in sera from breast cancer patients by proteomic analysis. Electrophoresis 2009, 30, 2168–2181. [Google Scholar] [CrossRef]
- Jiang, H.; Ma, N.; Shang, Y.; Zhou, W.; Chen, T.; Guan, D. Triosephosphate isomerase 1 suppresses growth, migration and invasion of hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 482, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yuan, S.; Hu, Y.; Zhang, H.; Wu, W.; Zeng, Z.; Yang, J.; Yun, J.; Xu, R.; Huang, P. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-bromopyruvate propyl ester. J. Bioenerg. Biomembr. 2012, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tang, Z.; Huang, A.; Chen, P.; Liu, P.; Yang, J.; Lu, W.; Liao, J.; Sun, Y.; Wen, S. Glyceraldehyde-3-phosphate dehydrogenase promotes cancer growth and metastasis through upregulation of SNAIL expression. Int. J. Oncol. 2017, 50, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, A.; Petrosino, M.; Ilari, A.; Pasquo, A.; Cipollone, A.; Maggi, M.; Chiaraluce, R.; Consalvi, V. The phosphoglycerate kinase 1 variants found in carcinoma cells display different catalytic activity and conformational stability compared to the native enzyme. PLoS ONE 2018, 13, e0199191. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Glatzle, J.; Bajaeifer, K.; Buhler, S.; Lehmann, T.; Konigsrainer, I.; Vollmer, J.P.; Sipos, B.; Ahmad, S.S.; Northoff, H.; et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 2013, 43, 586–590. [Google Scholar] [CrossRef]
- Ai, J.; Huang, H.; Lv, X.; Tang, Z.; Chen, M.; Chen, T.; Duan, W.; Sun, H.; Li, Q.; Tan, R.; et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell. Physiol. Biochem. 2011, 27, 207–216. [Google Scholar] [CrossRef]
- Daly, E.B.; Wind, T.; Jiang, X.M.; Sun, L.; Hogg, P.J. Secretion of phosphoglycerate kinase from tumour cells is controlled by oxygen-sensing hydroxylases. Biochim. Biophys. Acta 2004, 1691, 17–22. [Google Scholar] [CrossRef]
- Duan, Z.; Lamendola, D.E.; Yusuf, R.Z.; Penson, R.T.; Preffer, F.I.; Seiden, M.V. Overexpression of human phosphoglycerate kinase 1(PGK1) induces a multidrug resistance phenotype. Anticancer Res. 2002, 22, 1933–1941. [Google Scholar]
- Hwang, T.L.; Liang, Y.; Chien, K.Y.; Yu, J.S. Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics 2006, 6, 2259–2272. [Google Scholar] [CrossRef]
- Sun, S.; Liang, X.; Zhang, X.; Liu, T.; Shi, Q.; Song, Y.; Jiang, Y.; Wu, H.; Jiang, Y.; Lu, X.; et al. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. J. Cancer 2015, 112, 1332–1339. [Google Scholar] [CrossRef]
- Hitosugi, T.; Zhou, L.; Fan, J.; Arellano, M.; Khoury, H.J.; Lee, B.H.; Boggon, T.; Kang, S.; He, C.; Chen, J. Phosphoglycerate Mutase 1 Promotes Leukemia Metabolism by Coordinating Glycolysis and Biosynthesis, and Represents a Therapeutic Target in Leukemia Treatment. Blood 2012, 120, 860. [Google Scholar] [CrossRef]
- Liu, L.; Wang, S.; Zhang, Q.; Ding, Y. Identification of potential genes/proteins regulated by Tiam1 in colorectal cancer by microarray analysis and proteome analysis. Cell Biol. Int. 2008, 32, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Wu, H.; Lei, Y.; Zhang, H.; Liu, R.; Zhao, Y.; Chen, X.; Zeng, D.; Tong, A.; Chen, L.; et al. Quantitative proteomics identification of phosphoglycerate mutase 1 as a novel therapeutic target in hepatocellular carcinoma. Mol. Cancer 2010, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Buhrens, R.I.; Amelung, J.T.; Reymond, M.A.; Beshay, M. Protein expression in human non-small cell lung cancer: A systematic database. Pathobiology 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Durany, N.; Joseph, J.; Campo, E.; Molina, R.; Carreras, J. Phosphoglycerate mutase, 2,3-bisphosphoglycerate phosphatase and enolase activity and isoenzymes in lung, colon and liver carcinomas. Br. J. Cancer 1997, 75, 969–977. [Google Scholar] [CrossRef][Green Version]
- Zhang, D.; Wu, H.; Zhang, X.; Ding, X.; Huang, M.; Geng, M.; Li, H.; Xie, Z. Phosphoglycerate Mutase 1 Predicts the Poor Prognosis of Oral Squamous Cell Carcinoma and is Associated with Cell Migration. J. Cancer 2017, 8, 1943–1951. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jin, C.C.; Choi, J.E.; Hong, M.J.; Jung, D.K.; Do, S.K.; Baek, S.A.; Kang, H.J.; Kang, H.G.; Choi, S.H.; et al. Genetic polymorphisms in glycolytic pathway are associated with the prognosis of patients with early stage non-small cell lung cancer. Sci. Rep. 2016, 6, 35603. [Google Scholar] [CrossRef]
- Ejeskär, K.; Krona, C.; Carén, H.; Zaibak, F.; Li, L.; Martinsson, T.; Ioannou, P.A. Introduction of in vitro transcribed ENO1 mRNA into neuroblastoma cells induces cell death. BMC Cancer 2005, 5, 161. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, R.; Xue, Y.; Niu, Z.; Cui, K.; Yu, F.; Zhang, B.; Li, S. Role of enolase-1 in response to hypoxia in breast cancer: Exploring the mechanisms of action. Oncol. Rep. 2013, 29, 1322–1332. [Google Scholar] [CrossRef]
- Reece, K.M.; Richardson, E.D.; Cook, K.M.; Campbell, T.J.; Pisle, S.T.; Holly, A.J. Epidithiodiketopiperazines (ETPs) exhibit in vitro antiangiogenic and in vivo antitumor activity by disrupting the HIF-1α/p300 complex in a preclinical model of prostate cancer. Mol. Cancer 2014, 13, 91. [Google Scholar] [CrossRef]
- Chao, T.K.; Huang, T.S.; Liao, Y.P.; Huang, R.L.; Su, P.H.; Shen, H.Y.; Lai, H.C.; Wang, Y.C. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS ONE 2017, 12, e0182166. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Woolbright, B.L.; Pirani, K.; Didde, R.; Abbott, E.; Kaushik, G.; Martin, P.; Hamilton-Reeves, J.; Taylor, J.A., 3rd; Holzbeierlein, J.M. Tumor M2-PK: A novel urine marker of bladder cancer. PLoS ONE 2019, 14, e0218737. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.; Kumar, A.; Kumar, V.; Pollyea, D.A. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Jelinek, J.; Pastore, Y.D.; Guan, Y.; Prchal, J.F.; Prchal, J.T. Discrimination of polycythemias and thrombocytoses by novel, simple, accurate clonality assays and comparison with PRV-1 expression and BFU-E response to erythropoietin. Blood 2003, 101, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.S.; Prasad, M.H.; Reddy, P.P.; Rao, D.N. Leukocyte glucose 6 phosphate dehydrogenase as prognostic indicator in ANLL and CML. Indian J. Exp. Biol. 1997, 35, 155–158. [Google Scholar]
- Wilimas, J.A.; Dow, L.W.; Douglass, E.C.; Jenkins, J.J., 3rd; Jacobson, R.J.; Moohr, J.; Fialkow, P.J. Evidence for clonal development of Wilms’ tumor. Am. J. Pediatr. Hematol. Oncol. 1991, 13, 26–28. [Google Scholar] [CrossRef]
- Suh, S.; Kim, Y.H.; Goh, T.S.; Jeong, D.C.; Lee, C.S.; Jang, J.Y.; Cha, W.; Han, M.E.; Kim, S.J.; Kim, I.J.; et al. mRNA Expression of SLC5A5 and SLC2A Family Genes in Papillary Thyroid Cancer: An Analysis of The Cancer Genome Atlas. Yonsei Med. J. 2018, 59, 746–753. [Google Scholar] [CrossRef]
- Jonas, S.K.; Benedetto, C.; Flatman, A.; Hammond, R.H.; Micheletti, L.; Riley, C.; Riley, P.A.; Spargo, D.J.; Zonca, M.; Slater, T.F. Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br. J. Cancer 1992, 66, 185–191. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Chan, B. Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett. 2012, 586, 2389–2395. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Morris, H.P.; Weber, G. Behavior of transaldolase (EC 2.2.1.2) and transketolase (EC 2.2.1.1) Activities in normal, neoplastic, differentiating, and regenerating liver. Cancer Res. 1976, 36, 3189–3197. [Google Scholar] [PubMed]
- Yonashiro, R.; Eguchi, K.; Wake, M.; Takeda, N.; Nakayama, K. Pyruvate Dehydrogenase PDH-E1β Controls Tumor Progression by Altering the Metabolic Status of Cancer Cells. Cancer Res. 2018, 78, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mai, C.; Wei, Y.L.; Zhao, J.J.; Hu, Y.M.; Zeng, Z.L.; Yang, J.; Lu, W.H.; Xu, R.H.; Huang, P. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med. Oncol. 2013, 30, 552. [Google Scholar] [CrossRef] [PubMed]
- Juang, H.H. Modulation of mitochondrial aconitase on the bioenergy of human prostate carcinoma cells. Mol. Genet. Metab. 2004, 81, 244–252. [Google Scholar] [CrossRef]
- Tsui, K.H.; Feng, T.H.; Lin, Y.F.; Chang, P.L.; Juang, H.H. p53 downregulates the gene expression of mitochondrial aconitase in human prostate carcinoma cells. Prostate 2011, 71, 62–70. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Seki, M.; Hasegawa, D.; Kato, M.; Hyakuna, N.; Shuo, T.; Kimura, S.; Yoshida, K.; Kataoka, K.; Fujii, Y. Constitutional abnormalities of IDH1 combined with secondary mutations predispose a patient with Maffucci syndrome to acute lymphoblastic leukemia. Pediatr. Blood Cancer 2017, 64, 1–4. [Google Scholar] [CrossRef]
- Perrech, M.; Dreher, L.; Röhn, G.; Stavrinou, P.; Krischek, B.; Toliat, M.; Goldbrunner, R.; Timmer, M. Qualitative and Quantitative Analysis of IDH1 Mutation in Progressive Gliomas by Allele-Specific qPCR and Western Blot Analysis. Technol. Cancer Res. Treat. 2019, 1, 18. [Google Scholar] [CrossRef]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. IDH1-R132 changes vary according to NPM1 and other mutations status in AML. Leukemia 2019, 33, 1043–1047. [Google Scholar] [CrossRef]
- Van der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.J.; Corssmit, E.P.M.; Dinjens, W.N.; van de Horst-Schrivers, A.N.A.; Jansen, J.C.; de Jong, M.M.; Kunst, H.P.M.; Kusters, B. Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Lerario, A.M.; Everett, J.; Haymon, L.; Wham, D.; Mullane, M.; Wilson, T.L.; Rainville, I.; Rana, H.; Worth, A.J. Adrenocortical carcinoma and succinate dehydrogenase gene mutations: An observational case series. Eur. J. Endocrinol. 2017, 177, 439–444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stanley, K.; Friehling, E.; Davis, A.; Ranganathan, S. Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumor with SDHC Germline Mutation and Bilateral Renal and Neck Cysts. Pediatr. Dev. Pathol. 2019, 22, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.D.; Wang, Z.Y.; Chai, Y.C.; Wang, X.W.; Chen, D.Y.; Wu, H. Germline mutations and genotype-phenotype associations in head and neck paraganglioma patients with negative family history in China. Eur. J. Med. Genet. 2015, 58, 433–438. [Google Scholar] [CrossRef]
- Iwashita, H.; Okudela, K.; Matsumura, M.; Yamanaka, S.; Sawazumi, T.; Enaka, M.; Udaka, N.; Miyake, A.; Hibiya, T.; Miyake, N.; et al. Succinate dehydrogenase B-deficient renal cell carcinoma: A case report with novel germline mutation. Pathol. Int. 2017, 67, 585–589. [Google Scholar] [CrossRef]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G., Jr.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef]
- Muller, M.; Guillaud-Bataille, M.; Salleron, J.; Genestie, C.; Deveaux, S.; Slama, A.; de Paillerets, B.B.; Richard, S.; Benusiglio, P.R.; Ferlicot, S. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod. Pathol. 2018, 31, 974–983. [Google Scholar]
- Jiménez-Morales, S.; Pérez-Amado, C.J.; Langley, E.; Hidalgo-Miranda, A. Overview of mitochondrial germline variants and mutations in human disease: Focus on breast cancer (Review). Int. J. Oncol. 2018, 53, 923–936. [Google Scholar]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Schubert, S.; van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef]
- Lehtonen, H.J.; Kiuru, M.; Ylisaukko-Oja, S.K.; Salovaara, R.; Herva, R.; Koivisto, P.A.; Vierimaa, O.; Aittomäki, K.; Pukkala, E.; Launonen, V.; et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J. Med. Genet. 2006, 43, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tornmalm, J.; Widengren, J.; Vakifahmetoglu-Norberg, H.; Norberg, E. Characterization of the Role of the Malate Dehydrogenases to Lung Tumor Cell Survival. J. Cancer 2017, 8, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, C.S.; Hanson, W.R. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Brunengraber, H.; Roe, R.C. Anaplerotic molecules: Current and future. J. Inherit. Metab. Dis. 2006, 29, 327–331. [Google Scholar] [CrossRef]
- Jungas, R.L.; Halperin, M.L.; Brosnan, J.T. Quantitative analysis of amino acid oxidation and related gluconeogenesis in humans. Physiol. Rev. 1992, 72, 419–448. [Google Scholar] [CrossRef]
- Coller, H.A. Is cancer metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef]
- Moreadith, R.W.; Lehninger, A.L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria: Role of mitochondrial NAD(P)þ-dependent malic enzyme. J. Biol. Chem. 1984, 259, 6215–6221. [Google Scholar]
- Bauer, D.E.; Hatzivassilio, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavare, J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, J.; Kulkarni, S.; Zhang, W.; Gorka, J.E.; Mandel, J.A.; Goetzman, E.S.; Prochownik, E.V. Metabolic and oncogenic adaptations to pyruvate dehydrogenase inactivation in fibroblasts. J. Biol. Chem. 2019, 294, 5466–5486. [Google Scholar] [CrossRef] [PubMed]
- Beharry, Z.; Mahajan, S.; Zemskova, M. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Maciver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Plas, D.R.; Rathmell, J.C.; Fox, C.J.; Harris, M.H.; Thompson, C.B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 2001, 21, 5899–5912. [Google Scholar] [CrossRef]
- O’Neill, L.A. A broken Krebs cycle in macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1b through HIF-1a. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Rathmell, J.C. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab. 2013, 1, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Valle-Mendiola, A.; Soto-Cruz, I. Energetic metabolism and cancer. Vertientes 2014, 17, 108–113. [Google Scholar]
- Coloff, J.L.; Mason, E.F.; Altman, B.J.; Gerriets, V.A.; Liu, T.; Nichols, A.N.; Zhao, Y.; Wofford, J.A.; Jacobs, S.R.; Ilkayeva, O.; et al. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J. Biol. Chem. 2011, 286, 5921–5933. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Charm, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar]
- Mason, E.F.; Zhao, Y.; Goraksha-Hicks, P.; Coloff, J.L.; Gannon, H.; Jones, S.N.; Rathmell, J.C. Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl. Cancer Res. 2011, 70, 8066–8076. [Google Scholar] [CrossRef]
- Cornish, G.H.; Sinclair, L.V.; Cantrell, D.A. Differential regulation of T-cell growth by IL-2 and IL-15. Blood 2006, 108, 600–608. [Google Scholar] [CrossRef]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 Trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Goodman, T.M.; Zasadny, K.R.; Greenson, J.K.; Wahl, R.L. Expression of hexokinase II and Glut-1 in untreated human breast cancer. Nucl. Med. Biol. 2002, 29, 443–453. [Google Scholar] [CrossRef]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R.; et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.; McCracken, S.; Mak, T.W. Cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 299–311. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Dasu, A.; Toma-Dasu, I.; Karlsson, M. Theoretical simulation of tumour oxygenation and results from acute and chronic hypoxia. Phys. Med. Biol. 2003, 48, 2829–2842. [Google Scholar]
- Zeng, W.; Liu, P.; Pan, W.; Singh, S.R.; Wei, Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015, 356, 263–267. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Joskowicz, A.; Dulak, J. HIF-1 versus HIF-2- Is one more important than the other? Vascul. Pharmacol. 2012, 56, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohn, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Gen. Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 2007, 19, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 9, 651–662. [Google Scholar] [CrossRef]
- Lanning, N.J.; Carter-Su, C. Recent advances in growth hormone signaling. Rev. Endocr. Metab. Disord. 2006, 7, 225–235. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Chueh, F.Y.; Leong, K.F.; Yu, C.L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010, 402, 778–783. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Dorritie, K.A.; Redner, R.L.; Johnson, D.E. STAT transcription factors in normal stem cells. Adv. Biol. Regul. 2014, 56, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 mediates expression of autotoxin in breast cancer. PLoS ONE 2011, 6, e27851. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Pencik, J.; Pham, H.T.; Schomoeller, J.; Javaheri, T.; Schlenderer, M.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef]
- Carlsson, R.; Özen, I.; Barbariga, M.; Gaceb, A.; Roth, M.; Paul, G. STAT3 precedes HIF1α transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS ONE 2018, 13, e0194146. [Google Scholar] [CrossRef]
- Cui, Y.; Li, Y.Y.; Li, J.; Zhang, H.Y.; Wang, F.; Bai, X.; Li, S.S. STAT3 regulates hypoxia-induced epithelial mesenchymal transition in oesophageal squamous cell cancer. Oncol. Rep. 2016, 36, 108–116. [Google Scholar] [CrossRef]
- Rad, E.; Dodd, K.; Thomas, L.; Upadhyaya, M.; Tee, A. STAT3 and HIF1α signaling drives oncogenic cellular phenotypes in malignant peripheral nerve sheath tumors. Mol. Cancer Res. 2015, 13, 1149–1160. [Google Scholar] [CrossRef]
- Zheng, H.; Li, S.; Hsu, P.; Qu, C.K. Induction of a tumor-associated activating mutation in protein tyrosine phosphatase Ptpn11 (Shp2) enhances mitochondrial metabolism, leading to oxidative stress and senescence. J. Biol. Chem. 2013, 288, 25727–25738. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The important of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef]
- Elschami, M.; Scherr, M.; Philippens, B.; Gerardy-Schahn, R. Reduction of STAT3 expression induces mitochondrial dysfunction and autophagy in cardiac HL-1 cells. Eur. J. Cell Biol. 2013, 92, 21–29. [Google Scholar] [CrossRef]
- Xiong, H.; Su, W.Y.; Liang, Q.C.; Zhang, Z.G.; Chen, H.M.; Du, W.; Chen, Y.X.; Fang, J.Y. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Investig. 2009, 89, 717–725. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Weiss-Steider, B.; Rocha-Zavaleta, L.; Soto-Cruz, I. IL-2 enhances cervical cancer cells proliferation and JAK3/STAT5 phosphorylation at low doses, while at high doses IL-2 has opposite effects. Cancer Investig. 2014, 32, 115–125. [Google Scholar] [CrossRef]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef]
- Joung, J.H.; Lee, M.Y.; Lim, E.J.; Kim, M.S.; Hwang, T.S.; Kim, S.Y.; Ye, S.K.; Lee, J.D.; Park, T.; Woo, Y.S.; et al. Hypoxia activates the IGF-1 expression through STAT5b in human HepG2 cells. Biochem. Biophys. Res. Commun. 2007, 358, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Cholez, E.; Debuysscher, V.; Bourgeais, J.; Boudot, C.; Leprince, J.; Tron, F.; Brassart, B.; Regnier, A.; Bissac, E.; Pecnard, E.; et al. Evidence for a protective role of the STAT5 transcription factor against oxidative stress in human leukemic pre-B cells. Leukemia 2012, 26, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- White, U.A.; Coulter, A.A.; Miles, T.K.; Stephens, J.M. The STAT5A-mediated induction of pyruvate dehydrogenase kinase 4 expression by prolactin or growth hormone in adipocytes. Diabetes 2007, 56, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.Y.; Leong, K.F.; Cronk, R.J.; Venkitachalam, S.; Pabich, S.; Yu, C.L. Nuclear localization of pyruvate dehydrogenase complex-E2 (PDC-E2), a mitochondrial enzyme, and its role in signal transducer and activator of transcription 5 (STAT5)-dependent gene transcription. Cell. Signal. 2011, 23, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Fatrai, S.; Wierenga, A.T.; Daenen, S.M.; Vellenga, E.; Schuringa, J.J. Identification of HIF2 alpha as an important STAT5 target gene in human hematopoietic stem cells. Blood 2011, 24, 3320–3330. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene upregulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [CrossRef]
- Kaltenecker, D.; Themanns, M.; Mueller, K.M.; Spirk, K.; Golob-Schwarzl, N.; Friedbichler, K.; Kenner, L.; Haybaeck, J.; Moriggl, R. STAT5 deficiency in hepatocytes reduces diethylnitrosamine-induced liver tumorigenesis in mice. Cytokine 2019, 124, 154573. [Google Scholar] [CrossRef]
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinolog 2011, 152, 181–192. [Google Scholar] [CrossRef]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. N. Y. Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- Mason, J.I.; Keeney, D.S.; Bird, I.M. The regulation of 3 beta-hydroxysteroid dehydrogenase expression. Steroids 1997, 62, 164–168. [Google Scholar] [CrossRef]
- Abbaszade, I.G.; Clarke, T.R.; Park, C.H.; Payne, A.H. The mouse 3 beta-hydroxysteroid dehydrogenase multigene family includes two functionally distinct groups of proteins. Mol. Endocrinol. 1995, 9, 1214–1222. [Google Scholar] [PubMed]
- Holloway, M.G.; Cui, Y.; Laz, E.V.; Hosui, A.; Hennighausen, L.; Waxman, D.J. Loss of sexually dimorphic liver gene expression upon hepatocyte-specific deletion of Stat5a-Stat5b locus. Endocrinology 2007, 148, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.T.; Guo, Y.; Han, H.; Zhu, Y.; Bolli, R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2001, 98, 9050–9055. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, K.; Shi, W. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Frias, M.A.; Montessuit, C. JAK-STAT signaling and myocardial glucose metabolism. JAK STAT 2013, 2, e26458. [Google Scholar] [CrossRef][Green Version]
- Heusch, G.; Musiolik, J.; Kottenberg, E.; Peters, J.; Jakob, H.; Thielmann, M. STAT5 activation and cardioprotection by remote ischemic preconditioning in humans: Short communication. Circ. Res. 2012, 110, 111–115. [Google Scholar] [CrossRef]
- Manoochehri Khoshinani, H.; Afsahar, S.; Najafi, R. Hypoxia: A doublr-edge sword in cancer therapy. Cancer Investig. 2016, 34, 536–545. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors-similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Silver, I.A.; Hunt, T.K. Regulations of wound-healing angiogenesis-effect of oxygen gradients and inspired oxygen concentration. Surgery 1981, 90, 262–270. [Google Scholar] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lin, Q.; Glazer, P.M.; Yun, Z. Hypoxic tumor microenviroment and cancer cell diffentiation. Curr. Mol. Med. 2009, 9, 425–434. [Google Scholar] [CrossRef]
- Cárdenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008, 68, 5812–5819. [Google Scholar] [CrossRef]
- Kizaka-Kondoh, S.; Tanaka, S.; Harada, H.; Hiraoka, M. The HIF-1-active microenvironment: An environmental target for cancer therapy. Adv. Drug Deliv. Rev. 2009, 61, 623–632. [Google Scholar] [CrossRef]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia–inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression. Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2014, 356, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1 mediates expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.M.; Goodfriend, T.L.; Kaplan, N.O. Lactic dehydrogenase: Functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science 1964, 143, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Bacon, A.L.; Harris, A.L. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004, 36, 530–539. [Google Scholar] [CrossRef]
- Favaro, E.; Nardo, G.; Persano, L. Hypoxia inducible factor-1alpha inactivation unveils a link between tumor cell metabolism and hypoxia-induced cell death. Am. J. Pathol. 2008, 173, 1186–1201. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signaling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef]
- Koritzinsky, M.; Magagnin, M.G.; van den Beucken, T.; Seigneuric, R.; Savelkouls, K.; Dostie, J.; Pyronnet, S.; Kaufman, R.J.; Weppler, S.A.; Voncken, J.W.; et al. Gene expression during acute and prolongued hypoxia is regulated by distinct mechanism of translational control. EMBO J. 2006, 25, 1114–1125. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valle-Mendiola, A.; Soto-Cruz, I. Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers 2020, 12, 124. https://doi.org/10.3390/cancers12010124
Valle-Mendiola A, Soto-Cruz I. Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers. 2020; 12(1):124. https://doi.org/10.3390/cancers12010124
Chicago/Turabian StyleValle-Mendiola, Arturo, and Isabel Soto-Cruz. 2020. "Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes" Cancers 12, no. 1: 124. https://doi.org/10.3390/cancers12010124
APA StyleValle-Mendiola, A., & Soto-Cruz, I. (2020). Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers, 12(1), 124. https://doi.org/10.3390/cancers12010124