Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality

Abstract
1. Introduction
2. Hyperglycemia and Risk Factors for Cancer
2.1. Hyperglycemia and DNA Damage
2.2. Hyperglycemia and RNA
2.3. Hyperglycemia and Proteins
3. Hyperglycemia and Cancer Progression
3.1. Metabolic Reprogramming and Molecular Alterations
3.2. Avoiding Immune Destruction and Increasing Tumor Promoting Inflammation
3.3. Proliferation and Apoptosis Inhibition
3.4. Metastasis
4. Hyperglycemia and Treatment of Cancer
5. Hyperglycemia and Cancer Mortality
6. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heron, M. Deaths: Leading Causes for 2016. Natl. Vital Stat. Rep. 2018, 67, 1–77. [Google Scholar] [PubMed]
- Editorial. GLOBOCAN 2018: Counting the toll of cancer. Lancet 2018, 392, 985. [Google Scholar] [CrossRef]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Ma, Q. Hyperglycemia, a neglected factor during cancer progression. BioMed Res. Int. 2014, 2014, 461917. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.C.; Pollack, L.A. Therapy insight: Influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat. Clin. Pract. Oncol. 2005, 2, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Psaraikas, H.M. Clinical challenges in caring for patients with diabetes and cancer. Diabetes Spectr. 2006, 19, 157–162. [Google Scholar] [CrossRef][Green Version]
- Jacob, P.; Chowdhury, T.A. Management of diabetes in patients with cancer. QJM Int. J. Med. 2014, 108, 443–448. [Google Scholar] [CrossRef]
- Lega, I.C.; Austin, P.C.; Fischer, H.D.; Fung, K.; Krzyzanowska, M.K.; Amir, E.; Lipscombe, L.L. The impact of diabetes on breast cancer treatments and outcomes: A population-based study. Diabetes Care 2018, 41, 755–761. [Google Scholar] [CrossRef]
- Hershey, D.S.; Hession, S. Chemotherapy and Glycemic Control in Patients with Type 2 Diabetes and Cancer: A Comparative Case Analysis. Asia Pac. J. Oncol. Nurs. 2017, 4, 95–97. [Google Scholar] [CrossRef]
- Mouri, M.I.; Badireddy, M. Hyperglycemia; StatPearls Publishing: Treasure Island, FL, USA, 2019; pp. 1–5. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430900/ (accessed on 6 July 2019).
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef]
- Satija, A.; Spiegelman, D.; Giovannucci, E.; Hu, F.B. Type 2 diabetes and risk of cancer. BMJ 2015, 350. [Google Scholar] [CrossRef]
- Sieri, S.; Agnoli, C.; Pala, V.; Grioni, S.; Brighenti, F.; Pellegrini, N.; Masala, G.; Palli, D.; Mattiello, A.; Panico, S.; et al. Dietary glycemic index, glycemic load, and cancer risk: Results from the EPIC-Italy study. Sci. Rep. 2017, 7, 9757. [Google Scholar] [CrossRef] [PubMed]
- Sieri, S.; Muti, P.; Claudia, A.; Berrino, F.; Pala, V.; Grioni, S.; Abagnato, C.A.; Blandino, G.; Contiero, P.; Schunemann, H.J.; et al. Prospective study on the role of glucose metabolism in breast cancer occurrence. Int. J. Cancer 2012, 130, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Stattin, P.; Björ, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Noda, M.; Sawada, N.; Kato, M.; Hidaka, A.; Mizoue, T.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Sasazuki, S.; et al. High hemoglobin A1c levels within the non-diabetic range are associated with the risk of all cancers. Int. J. Cancer 2016, 138, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Dankner, R.; Chetrit, A.; Segal, P. Glucose tolerance status and 20 year cancer incidence. Isr. Med. Assoc. J. 2007, 9, 592–596. [Google Scholar] [PubMed]
- Meisel, P.; Dau, M.; Sümnig, W.; Holtfreter, B.; Houshmand, M.; Nauck, M.; Kocher, T. Association between glycemia, serum lipoproteins, and the risk of oral leukoplakia: The population-based Study of Health in Pomerania (SHIP). Diabetes Care 2010, 33, 1230–1232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.L.; Boniol, M.; Zheng, T.; Zhang, Y.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef]
- Barua, R.; Templeton, A.J.; Seruga, B.; Ocana, A.; Amir, E.; Ethier, J.L. Hyperglycemia and Survival in Solid Tumours: A Systematic Review and Meta-analysis. Clin. Oncol. 2018, 30, 215–224. [Google Scholar] [CrossRef]
- Basu, A.K. DNA damage, mutagenesis and cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Chan, J.C.N. Evidence for DNA damage as a biological link between diabetes and cancer. Chin. Med. J. 2015, 128, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Schröter, D.; Höhn, A. Role of Advanced Glycation End Products in Carcinogenesis and their Therapeutic Implications. Curr. Pharm. Des. 2019, 24, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.I.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, R.; Shuck, S.C.; Chan, Y.S.; Liu, X.; Bates, S.E.; Lim, P.P.; Tamae, D.; Lacoste, S.; O’Connor, T.R.; Termini, J. DNA advanced glycation end products (DNA-AGEs) are elevated in urine and tissue in an animal model of type 2 diabetes. Chem. Res. Toxicol. 2017, 30, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Simone, S.; Gorin, Y.; Velagapudi, C.; Abboud, H.E.; Habib, S.L. Mechanism of oxidative DNA damage in diabetes tuberin inactivation and downregulation of DNA repair enzyme 8-oxo-7,8-dihydro-2′-deoxyguanosine- DNA glycosylase. Diabetes 2008, 57, 2626–2636. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, F.; Leri, A.; Cesselli, D.; Limana, F.; Safai, B.; Nadal-Ginard, B.; Anversa, P.; Kajstura, J. Hyperglycemia Activates p53 and p53-Regulated Genes Leading to Myocyte Cell Death. Diabetes 2001, 50, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, C.-S.; Hou, G.-M.; Dong, M.-Z.; Wang, Z.-B.; Hou, Y.; Schatten, H.; Zhang, G.R.; Sun, Q.Y. Type 2 diabetes increases oocyte mtDNA mutations which are eliminated in the offspring by bottleneck effect. Reprod. Biol. Endocrinol. 2018, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, J.; Aoki, Y. Associations between hyperglycemia and somatic transversion mutations in mitochondrial DNA of people with diabetes mellitus. Diabetologia 2003, 46, 1559–1566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Y.; Zhang, X.; Gu, C.; Xia, J. Diabetes mellitus is associated with breast cancer: systematic review, meta-analysis, and in silico reproduction. Panminerva Med. 2015, 57, 101–108. [Google Scholar] [PubMed]
- Vairaktaris, E.; Kalokerinos, G.; Goutzanis, L.; Spyridonidou, S.; Vassiliou, S.; Derka, S.; Nkenke, E.; Yapijakis, C.; Vylliotis, A.; Lazaris, A.; et al. Diabetes alters expression of p53 and c-myc in different stages of oral oncogenesis. Anticancer Res. 2007, 27, 1465–1473. [Google Scholar] [PubMed]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Brownlee, M. Inhibition of GAPDH activity. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Tornovsky-Babeay, S.; Dadon, D.; Ziv, O.; Tzipilevich, E.; Kadosh, T.; Schyr-Ben Haroush, R.; Hija, A.; Stolovich-Rain, M.; Furth-Lavi, J.; Granot, Z.; et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in β cells. Cell Metab. 2014, 19, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Dai, X.; Ni, J.; Cao, N.; Yang, G.; Xue, J.; Wang, X. High concentration of sugars is genotoxic to folate-deficient cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2019, 814, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Parada, H., Jr.; Cleveland, R.J.; North, K.E.; Stevens, J.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M.; Martinez, M.E.; Gammon, M.D. Genetic polymorphisms of diabetes-related genes, their interaction with diabetes status, and breast cancer incidence and mortality: The Long Island Breast Cancer Study Project. Mol. Carcinog. 2019, 58, 436–446. [Google Scholar]
- Yuasa, K.; Aoki, N.; Hijikata, T. JAZF1 promotes proliferation of C2C12 cells, but retards their myogenic differentiation through transcriptional repression of MEF2C and MRF4-Implications for the role of Jazf1 variants in oncogenesis and type 2 diabetes. Exp. Cell Res. 2015, 336, 287–297. [Google Scholar] [CrossRef]
- Zhong, A.; Chang, M.; Yu, T.; Gau, R.; Riley, D.J.; Chen, Y.; Chen, P.-L. Aberrant DNA damage response and DNA repair pathway in high glucose conditions. J. Cancer Res. Updates 2018, 7, 64–74. [Google Scholar] [CrossRef]
- Yesil-Devecioglu, T.; Dayan, A.; Demirtunc, R.; Sardas, S. Role of DNA repair genes XRCC3 and XRCC1 in predisposition to type 2 diabetes mellitus and diabetic nephropathy. Endocrinol. Diabetes Y Nutr. 2019, 66, 90–98. [Google Scholar] [CrossRef]
- Ye, C.; Li, X.; Wang, Y.; Zhang, Y.; Cai, M.; Zhu, B.; Mu, P.; Xia, X.; Zhao, Y.; Weng, J.; et al. Diabetes causes multiple genetic alterations and downregulates expression of DNA repair genes in the prostate. Lab. Investig. 2011, 91, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Xavier, D.J.; Takahashi, P.; Evangelista, A.F.; Foss-Freitas, M.C.; Foss, M.C.; Donadi, E.A.; Passos, G.A.; Sakamoto-Hojo, E.T. Assessment of DNA damage and mRNA/miRNA transcriptional expression profiles in hyperglycemic versus non-hyperglycemic patients with type 2 diabetes mellitus. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 776, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.J.; Yim, H. The accumulation of DNA repair defects is the molecular origin of carcinogenesis. Tumor Biol. 2013, 34, 3293–3302. [Google Scholar] [CrossRef] [PubMed]
- Nissar, S.; Sameer, A.S.; Rasool, R.; Rashid, F. DNA repair gene—XRCC1 in relation to genome instability and role in colorectal carcinogenesis. Oncol. Res. Treat. 2014, 37, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Frisch, J.; Nagel, G.; Kraus, A.; Dörsam, B.; Thomas, A.D.; Reißig, S.; Waisman, A.; Kaina, B. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis 2015, 36, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Ji, H.; Pan, J.; He, P.; Zhou, X.; Zhang, H.; Zhou, Z.; Chen, Z. Downregulated XPA promotes carcinogenesis of bladder cancer via impairment of DNA repair. Tumor Biol. 2017, 39, 1–9. [Google Scholar] [CrossRef]
- Sha, Y.; Vartanian, V.; Owen, N.; Mengden Koon, S.J.; Calkins, M.J.; Thompson, C.S.; Mirafzali, Z.; Mir, S.; Goldsmith, L.E.; He, H.; et al. Modulation of UVB-induced Carcinogenesis by Activation of Alternative DNA Repair Pathways. Sci. Rep. 2018, 8, 705. [Google Scholar] [CrossRef] [PubMed]
- Merkel, P.; Khoury, N.; Bertolotto, C.; Perfetti, R. Insulin and glucose regulate the expression of the DNA repair enzyme XPD. Mol. Cell. Endocrinol. 2003, 201, 75–85. [Google Scholar] [CrossRef]
- Manoel-Caetano, F.S.; Xavier, D.J.; Evangelista, A.F.; Takahashi, P.; Collares, C.V.; Puthier, D.; Foss-Freitas, M.C.; Foss, M.C.; Donadi, E.A.; Passos, G.A.; et al. Gene expression profiles displayed by peripheral blood mononuclear cells from patients with type 2 diabetes mellitus focusing on biological processes implicated on the pathogenesis of the disease. Gene 2012, 511, 151–160. [Google Scholar] [CrossRef]
- Park, J.; Sarode, V.R.; Euhus, D.; Kittler, R.; Scherer, P.E. Neuregulin 1-HER axis as a key mediator of hyperglycemic memory effects in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 21058–21063. [Google Scholar] [CrossRef]
- Meng, J.; Feng, M.; Dong, W.; Zhu, Y.; Li, Y.; Zhang, P.; Wu, L.; Li, M.; Lu, Y.; Chen, H.; et al. Identification of HNF-4α as a key transcription factor to promote ChREBP expression in response to glucose. Sci. Rep. 2016, 6, 23944. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, W.H.; Azadzoi, K.M.; Dai, P.; Wang, Q.; Sun, J.B.; Zhang, W.T.; Shu, Y.; Yang, J.H.; Yan, Z. Alu RNA accumulation in hyperglycemia augments oxidative stress and impairs eNOS and SOD2 expression in endothelial cells. Mol. Cell. Endocrinol. 2016, 426, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Wu, X.; Chen, X.; Xu, H.; Zhang, T.; Xu, Y. Hypermethylated in cancer 1 (HIC1) mediates high glucose induced ROS accumulation in renal tubular epithelial cells by epigenetically repressing SIRT1 transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat. Commun. 2018, 9, 5228. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Price, C.; Li, Z.; Li, Y.; Cao, D.; Wiley, A.; He, C.; Gurbuxani, S.; Kunjamma, R.B.; Huang, H.; et al. miR-9 is an essential oncogenic microRNA specifically overexpressed in mixed lineage leukemia-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 11511–11516. [Google Scholar] [CrossRef] [PubMed]
- Al-Muhtaresh, H.A.; Al-Kafaji, G. Evaluation of Two-Diabetes Related microRNAs Suitability as Earlier Blood Biomarkers for Detecting Prediabetes and type 2 Diabetes Mellitus. J. Clin. Med. 2018, 7, 12. [Google Scholar] [CrossRef]
- Keßler, J.; Rot, S.; Bache, M.; Kappler, M.; Würl, P.; Vordermark, D.; Tauber, H.; Greither, T. miR-199a-5p regulates HIF-1α and OSGIN2 and its expression is correlated to soft-tissue sarcoma patients’ outcome. Oncol. Lett. 2016, 12, 5281–5288. [Google Scholar] [CrossRef]
- De Castro, J.P.W.; Blandino-Rosano, M.; Bernal-Mizrachi, E. The microRNA 199a Family Is Regulated by Glucose Levels in Pancreatic Beta Cells. Diabetes 2018, 67, 2170. [Google Scholar] [CrossRef]
- Lee, N.K.; Lee, J.H.; Ivan, C.; Ling, H.; Zhang, X.; Park, C.H.; Calin, G.A.; Lee, S.A. MALAT1 promoted invasiveness of gastric adenocarcinoma. BMC Cancer 2017, 17, 46. [Google Scholar] [CrossRef]
- Aird, J.; Baird, A.M.; Lim, M.C.J.; McDermott, R.; Finn, S.P.; Gray, S.G. Carcinogenesis in prostate cancer: The role of long non-coding RNAs. Non-Coding RNA Res. 2018, 3, 29–38. [Google Scholar] [CrossRef]
- Biswas, S.; Chakrabarti, S. Increased Extracellular Matrix Protein Production in Chronic Diabetic Complications: Implications of Non-Coding RNAs. Non-Coding RNA 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Tao, Z.F.; Li, X.M.; Zhang, H.; Yao, J.; Jiang, Q. Aberrant expression of long noncoding RNAs in early diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Feng, B.; Chakrabarti, S. ANRIL: A regulator of VEGF in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.; Yuan, Y.; Chen, C.; Zhou, J.; Wu, Y. The role of long non-coding RNA ANRIL in the carcinogenesis of oral cancer by targeting miR-125a. Biomed. Pharmacother. 2018, 103, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.T.; Xu, J.H.; Zheng, Z.R.; Zhao, Q.Q.; Zeng, X.S.; Cheng, S.X.; Liang, Y.H.; Hu, Q.F. Long non-coding RNA ANRIL promotes carcinogenesis via sponging miR-199a in triple-negative breast cancer. Biomed. Pharmacother. 2017, 96, 14–21. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Wang, D.D.; Du, C.-X.; Wang, Y. Long Noncoding RNA ANRIL Promotes Cervical Cancer Development by Acting as a Sponge of miR-186. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 26, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, M.; Niu, Q.; Zhang, F.; Yang, Y.; Jiang, X. Knockdown of long non-coding RNA ANRIL inhibits tumorigenesis in human gastric cancer cells via microRNA-99a-mediated down-regulation of BMI1. Braz. J. Med Biol. Res. 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Del Puerto-Nevado, L.; Minguez, P.; Corton, M.; Solanes-Casado, S.; Prieto, I.; Mas, S.; Sanz, A.B.; Gonzalez-Alonso, P.; Villaverde, C.; Portal-Nuñez, S.; et al. Molecular evidence of field cancerization initiated by diabetes in colon cancer patients. Mol. Oncol. 2019, 13, 857–872. [Google Scholar] [CrossRef]
- Sun, Q.; Li, J.; Gao, F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J. Diabetes 2017, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Bosma-Den Boer, M.M.; Van Wetten, M.L.; Pruimboom, L. Chronic inflammatory diseases are stimulated by current lifestyle: How diet, stress levels and medication prevent our body from recovering. Nutr. Metab. 2012, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Frazier, T.H.; DiBaise, J.K.; McClain, C.J. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. J. Parenter. Enter. Nutr. 2011, 35, 14S–20S. [Google Scholar] [CrossRef] [PubMed]
- Aeberli, I.; Gerber, P.A.; Hochuli, M.; Kohler, S.; Haile, S.R.; Gouni-Berthold, I.; Berthold, H.K.; Spinas, G.A.; Berneis, K. Low to moderate sugar-sweetened beverage consumption impairs glucose and lipid metabolism and promotes inflammation in healthy young men: A randomized controlled trial. Am. J. Clin. Nutr. 2011, 94, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.; Hancock, D.P.; Petocz, P.; Ceriello, A.; Brand-Miller, J. High-glycemic index carbohydrate increases nuclear factor-kappaB activation in mononuclear cells of young, lean healthy subjects1-3. Am. J. Clin. Nutr. 2008, 87, 1188–1193. [Google Scholar] [PubMed]
- Goyal, R.; Faizy, A.; Islam, N. Effect of hyperglycemia on inflammatory markers in patients with Type 2 Diabetes. Nat. Preced. 2008. [Google Scholar] [CrossRef]
- Akbari, M.; Hassan-Zadeh, V. Hyperglycemia Affects the Expression of Inflammatory Genes in Peripheral Blood Mononuclear Cells of Patients with Type 2 Diabetes. Immunol. Investig. 2018, 47, 654–665. [Google Scholar] [CrossRef]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef]
- Kumar, S.; Chan, C.J.; Coussens, L.M. Inflammation and Cancer. Encycl. Immunobiol. 2016, 4, 406–415. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Nakamura, T.; Ebihara, I.; Tomino, Y.; Koide, H. Alteration of growth-related proto-oncogene expression in diabetic glomeruli by a specific endothelin receptor A antagonist. Nephrol. Dial. Transplant. 2012, 11, 1528–1531. [Google Scholar] [CrossRef]
- Isoe, T.; Makino, Y.; Mizumoto, K.; Sakagami, H.; Fujita, Y.; Honjo, J.; Takiyama, Y.; Itoh, H.; Haneda, M. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 2010, 78, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Arumugam, A.; Joseph, R.; Monga, K.; Boopalan, T.; Agullo, P.; Gutierrez, C.; Nandy, S.; Subramani, R.; de la Rosa, J.M.; et al. Hyperglycemia enhances the proliferation of non-tumorigenic and malignant mammary epithelial cells through increased leptin/IGF1R signaling and activation of AKT/mTOR. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred-Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, G.; Zhang, X.; Li, R.; Wang, J.; Dong, Z.; He, Z. Increased phosphorylation of histone H3 at serine 10 is involved in Epstein-Barr virus latent membrane protein-1-induced carcinogenesis of nasopharyngeal carcinoma. BMC Cancer 2013, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, D.; Hamm-Künzelmann, B.; Brand, K. Glucose regulates the promoter activity of aldolase A and pyruvate kinase M2 via dephosphorylation of Sp1. FEBS Lett. 1997, 417, 325–328. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Lin, Y.C.; Lin, C.C.; Chen, W.M.; Chen, B.P.C.; Lee, H. High Glucose Induces VEGF-C Expression via the LPA 1/3-Akt-ROS-LEDGF Signaling Axis in Human Prostate Cancer PC-3 Cells. Cell. Physiol. Biochem. 2018, 50, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jia, X.; Duan, Y.; Xiao, H.; Sundqvist, K.G.; Permert, J.; Wang, F. Excess glucose induces hypoxia-inducible factor-1a in pancreatic cancer cells and stimulates glucose metabolism and cell migration. Cancer Biol. Ther. 2013, 14, 428–435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, W.; He, Q.; Yan, W.; Sun, J.; Chen, Z.; Liu, Z.; Lu, Z.; Hou, J.; Shao, Y.; Zhou, X.; et al. High glucose enhances the metastatic potential of tongue squamous cell carcinoma via the PKM2 pathway. Oncotarget 2017, 8, 111770–111779. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358.e9–371.e9. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaíba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.-E.; Kim, J.-Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [PubMed]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Rodriguez-Garcia, A.; Mayo, J.C.; Sainz, R.M. Regulation of GLUT transporters by flavonoids in androgen-sensitive and-insensitive prostate cancer cells. Endocrinology 2014, 155, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, L.A.; Calvo, M.B.; Figueroa, A.; Pulido, E.G.; Campelo, R.G. Potential role of sugar transporters in cancer and their relationship with anticancer therapy. Int. J. Endocrinol. 2010, 2010, 205357. [Google Scholar] [CrossRef]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Park, K.G. Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Grewer, C.; Otte, N.J.; Gameiro, A.; Albers, T.; Singh, K.; Shere, H.; Bonomi, M.; Holst, J.; Schlessinger, A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015, 11, e1004477. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef]
- Stoiber, K.; Nagło, O.; Pernpeintner, C.; Zhang, S.; Koeberle, A.; Ulrich, M.; Braig, S. Targeting de novo lipogenesis as a novel approach in anti-cancer therapy. Br. J. Cancer 2018, 118, 43–51. [Google Scholar] [CrossRef]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef]
- McCarthy, M.T.; Moncayo, G.; Hiron, T.K.; Jakobsen, N.A.; Valli, A.; Soga, T.; O’Callaghan, C.A. Purine nucleotide metabolism regulates expression of the human immune ligand MICA. J. Biol. Chem. 2018, 293, 3913–3924. [Google Scholar] [CrossRef]
- Elion, G.B. The purine path to chemotherapy. Science 1989, 244, 41–47. [Google Scholar] [CrossRef]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Desiderio, V. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.D.; Grothe, W.; Haerting, J.; Kleber, G.; Grothey, A.; Fleig, W.E. Chemotherapy in advanced gastric cancer: A systematic review and meta-analysis based on aggregate data. J. Clin. Oncol. 2006, 24, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [PubMed]
- Walling, J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Investig. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef] [PubMed]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Malvi, P.; Chaube, B.; Singh, S.V.; Mohammad, N.; Vijayakumar, M.V.; Singh, S.; Chouhan, S.; Bhat, M.K. Elevated circulatory levels of leptin and resistin impair therapeutic efficacy of dacarbazine in melanoma under obese state. Cancer Metab. 2018, 6, 2. [Google Scholar] [CrossRef]
- Cai, X.; Cao, C.; Li, J.; Chen, F.; Zhang, S.; Liu, B.; Zhang, W.; Zhang, X.; Ye, L. Inflammatory factor TNF & 3B1 promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF 3BA;B (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget 2017, 8, 58338–58352. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2002, 409, 307–312. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, S.; Sun, Y.; Matt, Y.; Colburn, N.H.; Shu, Y.; Han, X. JNK/AP-1 pathway is involved in tumor necrosis factor-α induced expression of vascular endothelial growth factor in MCF7 cells. Biomed. Pharmacother. 2009, 63, 429–435. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Stevens, M.; Wiley, J.W. Diabetic peripheral neuropathy: Evidence for apoptosis and associated mitochondrial dysfunction. Diabetes 2000, 49, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, N.R.; Stanczak, M.; Oliveira, I.; Filardy, A.; Silva, R.; Zippelius, A.; Laubli, H. Abstract LB-059: Hyperglycemia enhances cancer immune evasion by inducing alternative macrophage polarization through increased O-GlcNAcylation. Cancer Res. 2019, 79, LB-059. [Google Scholar] [CrossRef]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating Myeloid Derived Suppressor Cells (MDSC) That Accumulate in Premalignancy Share Phenotypic and Functional Characteristics With MDSC in Cancer. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Chu, Y. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-T.; Wu, J.-R.; Cheng, C.-C.; Wang, S.; Wang, H.-T.; Lee, M.-C.; Wu, W.-S. Reactive oxygen species-mediated PKC and integrin signaling promotes tumor progression of human hepatoma HepG2. Clin. Exp. Metastasis 2011, 28, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Venugopal, S.K.; Singh, U.; Jialal, I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-α and -β. Diabetes 2005, 54, 85–91. [Google Scholar] [CrossRef]
- Gonzalez, Y.; Herrera, M.T.; Soldevila, G.; Garcia-Garcia, L.; Fabian, G.; Perez-Armendariz, E.M.; Torres, M. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 2012, 13, 19. [Google Scholar] [CrossRef]
- Ho, M.-Y.; Tang, S.-J.; Chuang, M.-J.; Cha, T.-L.; Li, J.-Y.; Sun, G.-H.; Sun, K.-H. TNF-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3beta-dependent mechanism. Mol. Cancer Res. 2012, 10, 1109–1119. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Pearce, E.L. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Buttner, M.; Aigner, M.; Kreutz, M. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Kreutz, M. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Cosio, F.G. Effects of high glucose concentrations on human mesangial cell proliferation. J. Am. Soc. Nephrol. 1995, 5, 1600–1609. [Google Scholar] [PubMed]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH 2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell. Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Ning, H.; Qiu, X.; Baine, L.; Lin, G.; Lue, T.F.; Lin, C.S. Effects of high glucose on human cavernous endothelial cells. Urology 2012, 80, 1162.e7–1162.e11. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, S.; Singh, S.; Athavale, D.; Ramteke, P.; Pandey, V.; Joseph, J.; Mohan, R.; Shetty, P.K.; Bhat, M.K. Glucose induced activation of canonical Wnt signaling pathway in hepatocellular carcinoma is regulated by DKK4. Sci. Rep. 2016, 6, 27558. [Google Scholar] [CrossRef] [PubMed]
- Malvi, P.; Chaube, B.; Singh, S.V.; Mohammad, N.; Pandey, V.; Vijayakumar, M.V.; Radhakrishnan, R.M.; Vanuopadath, M.; Nair, S.S.; Nair, B.G.; et al. Weight control interventions improve therapeutic efficacy of dacarbazine in melanoma by reversing obesity-induced drug resistance. Cancer Metab. 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Ma, Q.; Li, J.; Liu, H.; Li, W.; Ma, G.; Xu, Q.; Zhou, S.; Wu, E. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS ONE 2011, 6, e27074. [Google Scholar] [CrossRef]
- Okumura, M.; Yamamoto, M.; Sakuma, H.; Kojima, T.; Maruyama, T.; Jamali, M.; Cooper, D.R.; Yasuda, K. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: Reciprocal involvement of PKC-α and PPAR expression. Biochim. Biophys. Acta Mol. Cell Res. 2002, 1592, 107–116. [Google Scholar] [CrossRef]
- Luo, J.; Xiang, Y.; Xu, X.; Fang, D.; Li, D.; Ni, F.; Zhu, X.; Chen, B.; Zhou, M. High Glucose-Induced ROS Production Stimulates Proliferation of Pancreatic Cancer via Inactivating the JNK Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 6917206. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yao, F.; Li, W.-H.; Wan, J.-N.; Zhang, Y.-M.; Tang, Z.; Khan, S.; Wang, C.H.; Sun, S.R. PKCδ-dependent Activation of the Ubiquitin Proteasome System is Responsible for High Glucose-induced Human Breast Cancer MCF-7 Cell Proliferation, Migration and Invasion. Asian Pac. J. Cancer Prev. 2014, 14, 5687–5692. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Youssef, A. Antidiabetic Drug Metformin Induces Apoptosis in Human MCF Breast Cancer via Targeting ERK Signaling. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2011, 19, 275–285. [Google Scholar] [CrossRef]
- Ding, C.-Z.; Guo, X.-F.; Wang, G.-L.; Wang, H.-T.; Xu, G.-H.; Liu, Y.-Y.; Wu, Z.J.; Chen, Y.H.; Wang, J.; Wang, W.G. High glucose contributes to the proliferation and migration of non-small-cell lung cancer cells via GAS5-TRIB3 axis. Biosci. Rep. 2018, 38, BSR20171014. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Qian, W.; Cheng, L.; Yan, B.; Han, L.; Xu, Q.; Ma, Q.; Ma, J. Hyperglycemia aggravates microenvironment hypoxia and promotes the metastatic ability of pancreatic cancer. Comput. Struct. Biotechnol. J. 2018, 16, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Kong, F.; Wu, X.; Ren, Y.; Wu, S.; Wu, K.; Jiang, Z.; Zhang, W. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the TGF-β 1 /PI3K/akt signaling pathway. Cell. Physiol. Biochem. 2015, 35, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Owens, P.; Gorska, A.E.; Chytil, A.; Ye, F.; Shi, C.; Weaver, V.M.; Kalluri, R.; Moses, H.L.; Novitskiy, S.V. Development of Aggressive Pancreatic Ductal Adenocarcinomas Depends on Granulocyte Colony Stimulating Factor Secretion in Carcinoma Cells. Cancer Immunol. Res. 2017, 5, 718–729. [Google Scholar] [CrossRef]
- Jian, Z.; Cheng, T.; Zhang, Z.; Raulefs, S.; Shi, K.; Steiger, K.; Kong, B. Glycemic Variability Promotes Both Local Invasion and Metastatic Colonization by Pancreatic Ductal Adenocarcinoma. CMGH 2018, 6, 429–449. [Google Scholar] [CrossRef]
- Nerlich, A.G.; Sauer, U.; Kolm-Litty, V.; Wagner, E.; Koch, M.; Schleicher, E.D. Expression of glutamine: Fructose-6-phosphate amidotransferase in human tissues: Evidence for high variability and distinct regulation in diabetes. Diabetes 1998, 47, 170–178. [Google Scholar] [CrossRef]
- Pandey, V.; Chaube, B.; Bhat, M.K. Hyperglycemia regulates MDR-1, drug accumulation and ROS levels causing increased toxicity of carboplatin and 5-fluorouracil in MCF-7 cells. J. Cell. Biochem. 2011, 112, 2942–2952. [Google Scholar] [CrossRef]
- Biernacka, K.M.; Uzoh, C.C.; Zeng, L.; Persad, R.A.; Bahl, A.; Gillatt, D.; Perks, C.M.; Holly, J.M. Hyperglycemia-induced chemoresistance of prostate cancer cells due to IGFBP2. Endocr. Relat. Cancer 2013, 20, 741–751. [Google Scholar] [CrossRef]
- Gerards, M.C.; van der Velden, D.L.; Baars, J.W.; Brandjes, D.P.M.; Hoekstra, J.B.L.; Vriesendorp, T.M.; Gerdes, V.E.A. Impact of hyperglycemia on the efficacy of chemotherapy—A systematic review of preclinical studies. Crit. Rev. Oncol./Hematol. 2017, 113, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, R.; Zhao, M.; Li, L.; Fan, L.; Che, X.M. High glucose promotes gastric cancer chemoresistance in vivo and in vitro. Mol. Med. Rep. 2015, 12, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Vishvakarma, N.K.; Kumar, A.; Singh, V.; Singh, S.M. Hyperglycemia of tumor microenvironment modulates stage-dependent tumor progression and multidrug resistance: Implication of cell survival regulatory molecules and altered glucose transport. Mol. Carcinog. 2013, 52, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Faria, M.C.; Santos, N.A.G.D.; Carvalho Rodrigues, M.A.; Rodrigues, J.L.; Barbosa Junior, F.; Santos, A.C. Dos. Effect of diabetes on biodistribution, nephrotoxicity and antitumor activity of cisplatin in mice. Chem. Biol. Interact. 2015, 229, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Jafar, N.; Edriss, H.; Nugent, K. The effect of short-term hyperglycemia on the innate immune system. Am. J. Med Sci. 2016, 351, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Hung, M.-C. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606.e7–620.e7. [Google Scholar] [CrossRef]
- Mu, Q.; Jiang, M.; Zhang, Y.; Wu, F.; Li, H.; Zhang, W.; Tang, D. Metformin inhibits proliferation and cytotoxicity and induces apoptosis via AMPK pathway in CD19-chimeric antigen receptor-modified T cells. Oncotargets Ther. 2018, 11, 1767–1776. [Google Scholar] [CrossRef]
- Svajger, U.; Jeras, M. Anti-inflammatory effects of resveratrol and its potential use in therapy of immune-mediated diseases. Int. Rev. Immunol. 2012, 31, 202–222. [Google Scholar] [CrossRef] [PubMed]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer Molecular Mechanisms of Resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Soto, B.L.; Hank, J.A.; Van De Voort, T.J.; Subramanian, L.; Polans, A.S.; Rakhmilevich, A.L.; Sondel, P.M. The anti-tumor effect of resveratrol alone or in combination with immunotherapy in a neuroblastoma model. Cancer Immunol. Immunother. 2011, 60, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Singh, N.P.; Singh, U.P.; Nagarkatti, P.S.; Nagarkatti, M. Resveratrol prevents endothelial cells injury in high-dose interleukin-2 therapy against melanoma. PLoS ONE 2012, 7, e35650. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Demark-Wahnefried, W.; Peterson, C.M.; Norian, L.A. Targeting Glucose Metabolism to Enhance Immunotherapy: Emerging Evidence on Intermittent Fasting and Calorie Restriction Mimetics. Front. Immunol. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Duan, Q.; Li, H.; Gao, C.; Zhao, H.; Wu, S.; Wu, H.; Yin, T. High glucose promotes pancreatic cancer cells to escape from immune surveillance via AMPK-Bmi1-GATA2-MICA/B pathway. J. Exp. Clin. Cancer Res. 2019, 38, 192. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef]
- Zhao, J.; Zeng, D.; Liu, Y.; Luo, Y.; Ji, S.; Li, X.; Chen, T. Selenadiazole derivatives antagonize hyperglycemia-induced drug resistance in breast cancer cells by activation of AMPK pathways. Metallomics 2019, 9, 535–545. [Google Scholar] [CrossRef]
- Zeng, L.; Biernacka, K.M.; Holly, J.M.P.; Jarrett, C.; Morrison, A.A.; Morgan, A.; Winters, Z.E.; Foulstone, E.J.; Shield, J.P.; Perks, C.M. Hyperglycemia confers resistance to chemotherapy on breast cancer cells: The role of fatty acid synthase. Endocr. Relat. Cancer 2010, 17, 539–551. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Yang, D.Q.; Guo, Z.; Potter, D.A.; Cleary, M.P. Metformin Treatment for the Prevention and/or Treatment of Breast/Mammary Tumorigenesis. Curr. Pharmacol. Rep. 2015, 1, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Monzavi-Karbassi, B.; Gentry, R.; Kaur, V.; Siegel, E.R.; Jousheghany, F.; Medarametla, S.; Fuhrman, B.J.; Safar, A.M.; Hutchins, L.F.; Kieber-Emmons, T. Pre-diagnosis blood glucose and prognosis in women with breast cancer. Cancer Metab. 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Garza, C.; Shaw-Dulin, R.; Lara-Medina, F.; Bacon, L.; Rivera, D.; Urzua, L.; Aguila, C.; Ramirez-Morales, R.; Santamaria, J.; Bargallo, E.; et al. Impact of Diabetes and Hyperglycemia on Survival in Advanced Breast Cancer Patients. Exp. Diabetes Res. 2012, 2012, 732027. [Google Scholar] [CrossRef]
- Vissers, P.A.J.; Falzon, L.; van de Poll-Franse, L.V.; Pouwer, F.; Thong, M.S.Y. The impact of having both cancer and diabetes on patient-reported outcomes: A systematic review and directions for future research. J. Cancer Surviv. 2016, 10, 406–415. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
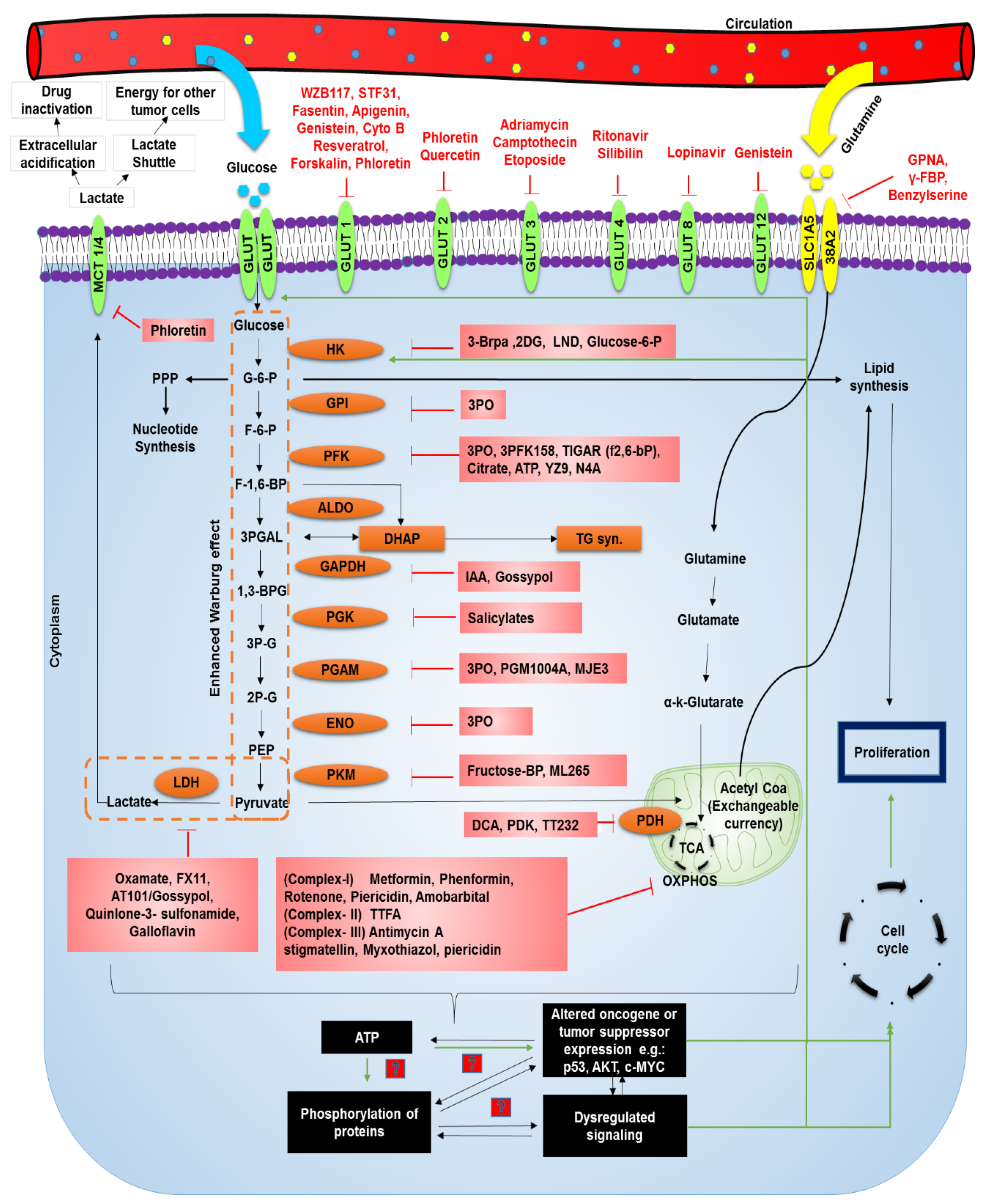{kind=link}
| Target | Agent | Cancer | Phase | ReferenceNCT No./Pubmed ID |
|---|---|---|---|---|
| GLUT1 | WZB117 | NSCLC | H1299 and A549 (in vitro and in vivo) | 22689530 |
| Breast | MCF-7 (in vitro and in vivo) | |||
| Breast | MCF-7/ADR resistant (in vitro) | 28609310 | ||
| Breast | MDA-MB-231 and MCF-7 | 27011212 | ||
| Colon | 5-FU-resistant human colon cancer cell line (in vitro) | 25227787 | ||
| Neuroblastoma | SH-SY5Y (in vitro) | 30553996 | ||
| Glioblastoma | Tumor-derived A172 | 29949049 | ||
| STF31 | RCC | RCC4, Caki-1, SN12C (in vitro) 786-O and (in vitro and in vivo) | 21813754 | |
| Apigenin | Pancreatic cancer | CD18 and S2-013 pancreatic cancer cell lines | 18953257 | |
| Colon cancer | Phase 2 prevention of the recurrence of neoplasia | NCT00609310 | ||
| Genistein/Isoflavone G-2535 | Hepatocellular carcinoma | HCC-LM3, SMMC7721, Hep3B, Bel-7402, and Huh-7 | 28926527 | |
| Prostate | Phase 2 | NCT00058266 | ||
| NSCLC | Phase 2 | NCT01628471 | ||
| Colorectal | Phase 2 | NCT01985763 | ||
| Prostate | Phase 3 | NCT00584532 | ||
| Breast | Phase 2 | NCT00290758 | ||
| Endometrial | Phase 1 | NCT00099008 | ||
| Pancreatic | Phase 2 | NCT00882765 | ||
| Bladder (I, II, III) | Phase 2 | NCT00118040 | ||
| Kidney | Early Phase 1 | NCT00276835 | ||
| Melanoma | Early Phase 1 | NCT00276835 | ||
| Head and Neck | Phase 1 | NCT02075112 | ||
| Leukemia | Phase 1 | NCT00004858 | ||
| Lymphoma | Phase 1 | NCT00004858 | ||
| Resveratrol/SRT501 | Ovarian cancer | PA-1, OVCAR3, MDAH2774,SKOV-3 | 25307508 | |
| Colon | Phase 1 | NCT00256334 | ||
| Liver | Phase 2 | NCT02261844 | ||
| Colon | Phase 1 | NCT00433576 | ||
| Colorectal | Phase 1 | NCT00920803 | ||
| Solid Tumor | Phase 1 | NCT00098969 | ||
| Multiple Myeloma | Phase 2 | NCT00920556 | ||
| Forskolin | Multiple Myeloma | H929 and OM-2 | 26306624 | |
| Quercetin | Breast and ovarian cancer | MCF-7, MDA-MB-231, HBL100, BT549, OVCAR5, TOV112D, OVCAR3, CAOV3 | 26259240 | |
| GLUT2 | Phloretin | Hepatocellular carcinoma | HepG2 | 19123483 |
| GLUT3 | Adriamycin and etoposide | Cervical and colon cancer | Hela and Caco-2 cell lines in vitro and in vivo | 20870738 |
| GLUT4 | Ritonavir | Multiple myeloma | MM.1S and U266 cell lines | 22452979 |
| GLUT5 | N-[4-(methylsulfonyl)-2-nitrophenyl]-1,3-benzodioxol-5-amine (MSNBA) | Breast cancer | MCF-7 | 27074918 |
| SLCA15 | GPNA, Benzylserine, γ-FBP, AOC, Chloroalanine | in silico | 26444490, 29212300 | |
| HK | 3-Bromopyruvate | Melanoma | in vivo | 30206027 |
| 2 Deoxy Glucose | Breast cancer | SKBR-3, MCF-7, MDA-MB-468, BT474 | 12232767 | |
| Prostate cancer | Phase 2 | NCT00633087 | ||
| Solid tumors | Phase 1 | NCT00096707 | ||
| Lonidamine | Melanoma | DB-1 xenograft model | 27497601 | |
| 3PO | Bladder cancer | in vitro | 26504012 | |
| PFK | N4A | Lung cancer, colon cancer, pancreatic cancer | in vivo | 23674815 |
| Breast Cancer | in vivo | 18202014 | ||
| Breast Cancer, Cervical cancer | HeLa, T47D | 21957443 | ||
| GAPDH | Gossypol | Non-small Cell Lung Cancer | in vitro | 30038571 |
| Non-small Cell Lung Cancer | in vitro | 31055235 | ||
| PKM | ML265 | Lung cancer | in vitro | 23905203 |
| LDH | Oxamate | Renal cell carcinoma | in vivo | 28983605 |
| FX11 | Breast Cancer | in vivo | 28243322 | |
| Neuroblastoma | in vitro | 27919448 | ||
| Prostate cancer | in vitro | 25983002 | ||
| AT101 /Gossypol | Lymphoma | in vivo | 20133848 | |
| Prostate Cancer | Phase 2 | NCT00666666 | ||
| Small Cell Lung Cancer | Phase 2 | NCT00773955 | ||
| Galloflavin | Breast cancer | in vitro | 22954722 | |
| Endometrial cancer | in vitro | 25631326 | ||
| PDH | DCA | Oral squamous cell carcinoma | in vitro | 25544754 |
| Head and Neck cancer | Phase 1 | NCT01163487 | ||
| TT232 | Melanoma | Phase 2 | 16393913 | |
| MCT-4 | Phloretin | Breast cancer, prostate cancer, lymphoma | in vitro | 27127175 |
| Complex 1 | Metformin | Colon cancer | in vitro and in vivo | 24843020 |
| Rotenone | Leukemia | in vitro | 12496265 | |
| Piericidin | Breast cancer | in vitro | 23690779 | |
| Complex 2 | TTFA | Melanoma | in vitro | 26521302 |
| Complex 3 | Stigmatellin | Lung cancer and Bone osteosarcoma | in vitro | 17562787 |
| Myxothiazol | Colon cancer | in vitro | 24772329 |
| Cancer | Combination | Phase | NCT No: |
|---|---|---|---|
| Metastatic colorectal cancer | Metformin, 5-Fluorouracil | Phase 2 | NCT01941953 |
| Her2 positive breast cancer | Liposomal Doxorubicin, Docetaxel, Trastuzumab, Metformin | Phase 2 | NCT02488564 |
| Breast cancer | Metformin, Doxorubicin | Phase 2 | NCT02472353 |
| Human epidermal growth factor 2 negative carcinoma of breast | Metformin, Myocet, Cyclophosphamide | Phase 2 | NCT01885013 |
| Myocet + Cyclophosphamide | |||
| Diffuse large B-cell lymphoma | Metformin, Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, Prednisone, Pegfilgrastine | Phase 2 | NCT02531308 |
| Lung cancer, Breast cancer, Pancreatic cancer, Head and neck cancer, Gastric cancer | 2-Deoxyglucose, Docetaxel | Phase 1 | NCT00096707 |
| Pancreatic cancer | Capecitabine, Cisplatin, Epirubicin, Gemcitabine, Metformin | Phase 2 | NCT01167738 |
| Prostate cancer | Rosiglitazone and Placebo | Phase 3 | NCT00182052 |
| Pancreatic cancer | Pioglitazone | Phase 2 | NCT01838317 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers 2019, 11, 1402. https://doi.org/10.3390/cancers11091402
Ramteke P, Deb A, Shepal V, Bhat MK. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers. 2019; 11(9):1402. https://doi.org/10.3390/cancers11091402
Chicago/Turabian StyleRamteke, Pranay, Ankita Deb, Varsha Shepal, and Manoj Kumar Bhat. 2019. "Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality" Cancers 11, no. 9: 1402. https://doi.org/10.3390/cancers11091402
APA StyleRamteke, P., Deb, A., Shepal, V., & Bhat, M. K. (2019). Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers, 11(9), 1402. https://doi.org/10.3390/cancers11091402