The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Characteristics of Pyroptosis
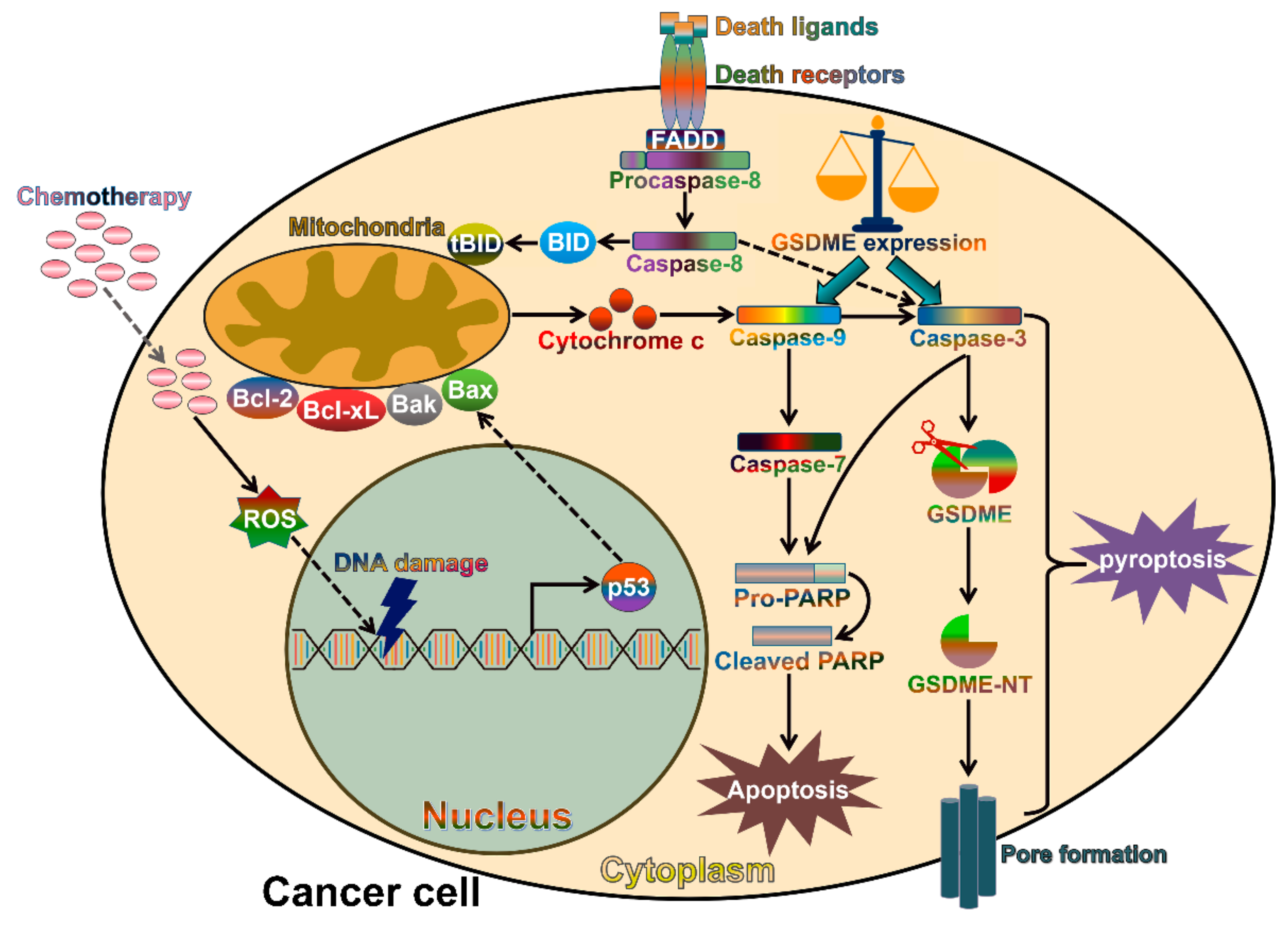
3. The Canonical Pyroptosis Pathway
4. The Non-Canonical Pyroptosis Pathway
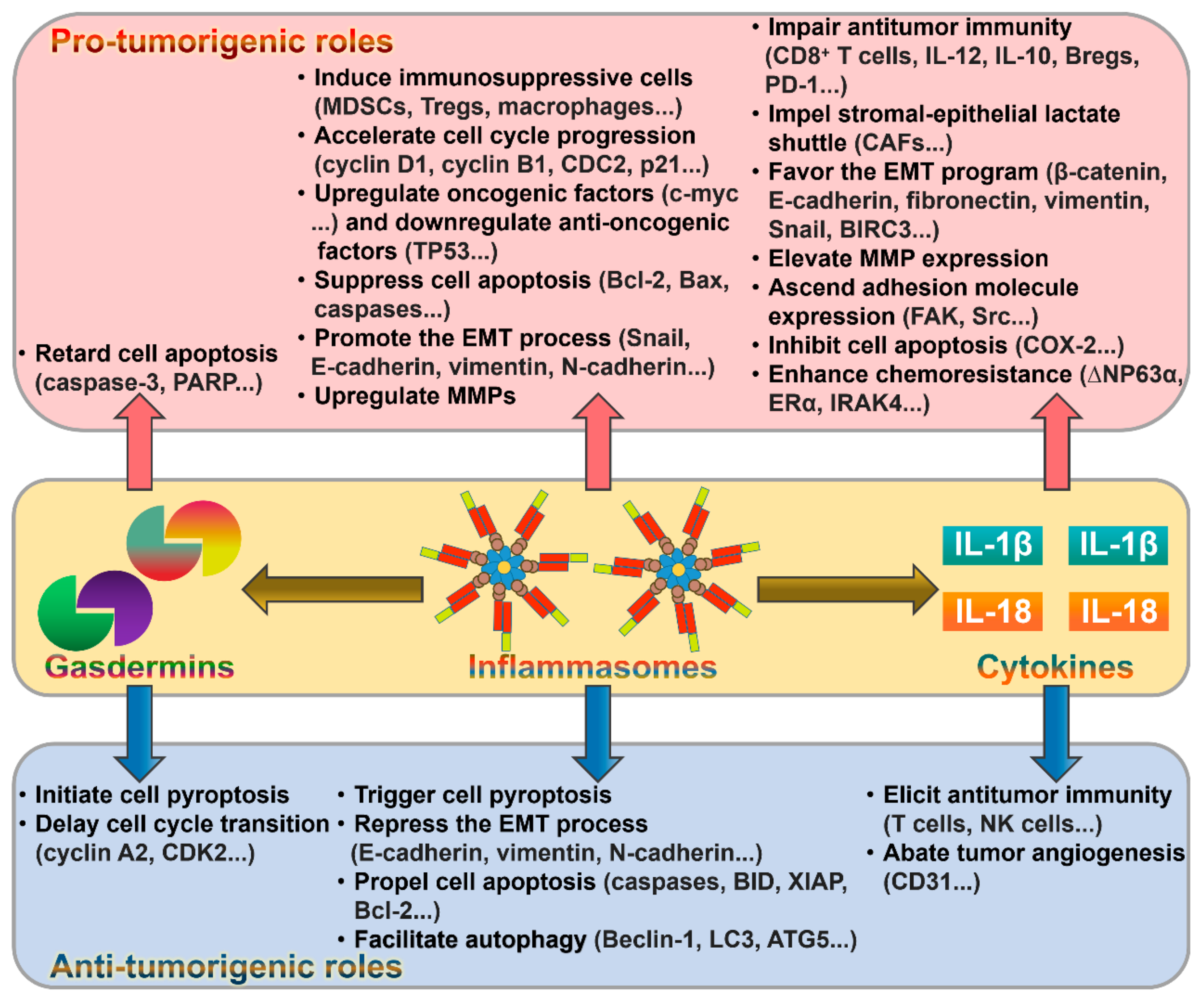
5. The Role of Inflammasomes in Cancer
6. The Contribution of Gasdermins to Cancer Progression
7. The Emerging Role of Inflammasome-Dependent Cytokines in Cancer Pathogenesis
7.1. IL-1β and Cancer
7.2. Therapeutic Potential of IL-1 Neutralization in Cancer
7.3. IL-18 and Cancer
7.4. Potential Efficacy of Anti-IL-18 in Cancer Therapy
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- He, Y.; Amer, A.O. Microbial Modulation of Host Apoptosis and Pyroptosis. Front. Cell Infect. Microbiol. 2014, 4, 83. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.D. Inflammatory Cell Death in Intestinal Pathologies. Immunol. Rev. 2017, 280, 57–73. [Google Scholar] [CrossRef]
- Fearnhead, H.O.; Rodriguez, J.; Govek, E.E.; Guo, W.; Kobayashi, R.; Hannon, G.; Lazebnik, Y.A. Oncogene-dependent Apoptosis is Mediated by Caspase-9. Proc. Natl. Acad. Sci. USA 1998, 95, 13664–13669. [Google Scholar] [CrossRef]
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent Membrane Repair Negatively Regulates Pyroptosis Downstream of GSDMD Activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Gong, Y.N.; Guy, C.; Crawford, J.C.; Green, D.R. Biological Events and Molecular Signaling following MLKL Activation during Necroptosis. Cell Cycle 2017, 16, 1748–1760. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Hornung, V. Pore Formation by GSDMD is the Effector Mechanism of Pyroptosis. EMBO J. 2016, 35, 2167–2169. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Gasdermin D: The Long-awaited Executioner of Pyroptosis. Cell Res. 2015, 25, 1183–1184. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Zheng, Y.; Jiang, J.; Hu, J. What role does pyroptosis play in microbial infection? J. Cell. Physiol. 2019, 234, 7885–7892. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhang, C.C.; Li, C.G.; Wang, Y.F.; Xu, L.H.; He, X.H.; Zeng, Q.Z.; Zeng, C.Y.; Mai, F.Y.; Hu, B.; Ouyang, D.Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Hornung, V. Of inflammasomes and pathogens—Sensing of microbes by the inflammasome. EMBO Mol. Med. 2013, 5, 814–826. [Google Scholar] [CrossRef]
- Stowe, I.; Lee, B.; Kayagaki, N. Caspase-11: Arming the guards against bacterial infection. Immunol. Rev. 2015, 265, 75–84. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Sharma, N.; Jha, S. NLR-regulated pathways in cancer: Opportunities and obstacles for therapeutic interventions. Cell. Mol. Life Sci. CMLS 2016, 73, 1741–1764. [Google Scholar] [CrossRef]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef]
- Caneparo, V.; Landolfo, S.; Gariglio, M.; De Andrea, M. The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front. Immunol. 2018, 9, 1180. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef]
- Lee, J.; Li, L.; Gretz, N.; Gebert, J.; Dihlmann, S. Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and -independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene 2012, 31, 1242–1253. [Google Scholar] [CrossRef]
- Yu, J.W.; Wu, J.; Zhang, Z.; Datta, P.; Ibrahimi, I.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249. [Google Scholar] [CrossRef]
- De Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrin, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef]
- Garib, F.Y.; Rizopulu, A.P.; Kuchmiy, A.A.; Garib, V.F. Inactivation of Inflammasomes by Pathogens Regulates Inflammation. Biochem. Biokhimiia 2016, 81, 1326–1339. [Google Scholar] [CrossRef]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638. [Google Scholar] [CrossRef]
- Hellmich, K.A.; Levinsohn, J.L.; Fattah, R.; Newman, Z.L.; Maier, N.; Sastalla, I.; Liu, S.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 2012, 7, e49741. [Google Scholar] [CrossRef]
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Komuves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542. [Google Scholar] [CrossRef]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015, 6, 7827. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Magnotti, F.; Belot, A.; Henry, T. The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018, 76, fty020. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis—A cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Ruhl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- Monteleone, M.; Stow, J.L.; Schroder, K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine 2015, 74, 213–218. [Google Scholar] [CrossRef]
- Monteleone, M.; Stanley, A.C.; Chen, K.W.; Brown, D.L.; Bezbradica, J.S.; von Pein, J.B.; Holley, C.L.; Boucher, D.; Shakespear, M.R.; Kapetanovic, R.; et al. Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 2018, 24, 1425–1433. [Google Scholar] [CrossRef]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1beta Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef]
- Di Virgilio, F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol. Rev. 2013, 65, 872–905. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Chen, M.; Wang, H.; Chen, W.; Meng, G. Regulation of adaptive immunity by the NLRP3 inflammasome. Int. Immunopharmacol. 2011, 11, 549–554. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. CMLS 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Huang, C.F.; Chen, L.; Li, Y.C.; Wu, L.; Yu, G.T.; Zhang, W.F.; Sun, Z.J. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 116. [Google Scholar] [CrossRef]
- Duan, S.; Wang, N.; Huang, L.; Shao, H.; Zhang, P.; Wang, W.; Wu, Y.; Wang, J.; Liu, H.; Zhang, Q.; et al. NLRP3 inflammasome activation involved in LPS and coal tar pitch extract-induced malignant transformation of human bronchial epithelial cells. Environ. Toxicol. 2019, 34, 585–593. [Google Scholar] [CrossRef]
- Huang, L.; Duan, S.; Shao, H.; Zhang, A.; Chen, S.; Zhang, P.; Wang, N.; Wang, W.; Wu, Y.; Wang, J.; et al. NLRP3 deletion inhibits inflammation-driven mouse lung tumorigenesis induced by benzo(a)pyrene and lipopolysaccharide. Respir. Res. 2019, 20, 20. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Zang, Y.; Wang, F. NLRP3 inflammasome inactivation driven by miR2233p reduces tumor growth and increases anticancer immunity in breast cancer. Mol. Med. Rep. 2019, 19, 2180–2188. [Google Scholar] [CrossRef]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Hua, M.; Wang, R.; Zhong, C.; Yu, J.; Han, F.; He, N.; Zhao, Y.; Liu, G.; et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 2017, 8, 108571–108583. [Google Scholar] [CrossRef]
- Wei, Q.; Zhu, R.; Zhu, J.; Zhao, R.; Li, M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol. Res. 2019, 27, 827–834. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef]
- Deng, Q.; Geng, Y.; Zhao, L.; Li, R.; Zhang, Z.; Li, K.; Liang, R.; Shao, X.; Huang, M.; Zuo, D.; et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 2019, 442, 21–30. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Du, Q.; Lu, P.; Fan, H.; Lu, J.; Hu, R. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp. Cell Res. 2016, 342, 184–192. [Google Scholar] [CrossRef]
- Hu, H.; Wang, Y.; Ding, X.; He, Y.; Lu, Z.; Wu, P.; Tian, L.; Yuan, H.; Liu, D.; Shi, G.; et al. Long non-coding RNA XLOC_000647 suppresses progression of pancreatic cancer and decreases epithelial-mesenchymal transition-induced cell invasion by down-regulating NLRP3. Mol. Cancer 2018, 17, 18. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef]
- Yin, X.F.; Zhang, Q.; Chen, Z.Y.; Wang, H.F.; Li, X.; Wang, H.X.; Li, H.X.; Kang, C.M.; Chu, S.; Li, K.F.; et al. NLRP3 in human glioma is correlated with increased WHO grade, and regulates cellular proliferation, apoptosis and metastasis via epithelial-mesenchymal transition and the PTEN/AKT signaling pathway. Int. J. Oncol. 2018, 53, 973–986. [Google Scholar] [CrossRef]
- Zou, J.; Yang, Y.; Yang, Y.; Liu, X. Polydatin suppresses proliferation and metastasis of non-small cell lung cancer cells by inhibiting NLRP3 inflammasome activation via NF-kappaB pathway. Biomed. Pharmacother. 2018, 108, 130–136. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef]
- Hu, Q.; Zhao, F.; Guo, F.; Wang, C.; Fu, Z. Polymeric Nanoparticles Induce NLRP3 Inflammasome Activation and Promote Breast Cancer Metastasis. Macromol. Biosci. 2017, 17, 1700273. [Google Scholar] [CrossRef]
- Shang, S.; Wang, L.; Zhang, Y.; Lu, H.; Lu, X. The Beta-Hydroxybutyrate Suppresses the Migration of Glioma Cells by Inhibition of NLRP3 Inflammasome. Cell. Mol. Neurobiol. 2018, 38, 1479–1489. [Google Scholar] [CrossRef]
- Wang, Y.C.; Li, W.Z.; Wu, Y.; Yin, Y.Y.; Dong, L.Y.; Chen, Z.W.; Wu, W.N. Acid-sensing ion channel 1a contributes to the effect of extracellular acidosis on NLRP1 inflammasome activation in cortical neurons. J. Neuroinflamm. 2015, 12, 246. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerback, C.; Rosdahl, I.; Soderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamai, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverenyi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef]
- Lee, B.L.; Mirrashidi, K.M.; Stowe, I.B.; Kummerfeld, S.K.; Watanabe, C.; Haley, B.; Cuellar, T.L.; Reichelt, M.; Kayagaki, N. ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 2018, 8, 3788. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Flavell, R.A. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle 2011, 10, 1936–1939. [Google Scholar] [CrossRef]
- Papin, S.; Cuenin, S.; Agostini, L.; Martinon, F.; Werner, S.; Beer, H.D.; Grutter, C.; Grutter, M.; Tschopp, J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007, 14, 1457–1466. [Google Scholar] [CrossRef]
- Richards, N.; Schaner, P.; Diaz, A.; Stuckey, J.; Shelden, E.; Wadhwa, A.; Gumucio, D.L. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 2001, 276, 39320–39329. [Google Scholar] [CrossRef]
- Sharma, D.; Malik, A.; Guy, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.D. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 2018, 154, 948–964.e8. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, X.; Yang, X.; Wan, D.; Sun, L.; Gu, M.; Li, M.; Zhu, Z.; Wang, J.; Shang, Z.; et al. Expression and clinical significance of absent in melanoma 2 in colorectal cancer. Biomed. Pharmacother. 2017, 94, 843–849. [Google Scholar] [CrossRef]
- Martinez-Cardona, C.; Lozano-Ruiz, B.; Bachiller, V.; Peiro, G.; Algaba-Chueca, F.; Gomez-Hurtado, I.; Such, J.; Zapater, P.; Frances, R.; Gonzalez-Navajas, J.M. AIM2 deficiency reduces the development of hepatocellular carcinoma in mice. Int. J. Cancer 2018, 143, 2997–3007. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, C.; Yang, Y.; Wang, K.; Zhou, Y.; Zhou, Y.; Wang, R.; Li, T.; Hu, R. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J. Cell. Physiol. 2019, 234, 20161–20173. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, M.; Ying, Q.; Xie, X.; Yue, S.; Tong, B.; Wei, Q.; Bai, Z.; Ma, L. Decrease of AIM2 mediated by luteolin contributes to non-small cell lung cancer treatment. Cell Death Dis. 2019, 10, 218. [Google Scholar] [CrossRef]
- Farshchian, M.; Nissinen, L.; Siljamaki, E.; Riihila, P.; Piipponen, M.; Kivisaari, A.; Kallajoki, M.; Grenman, R.; Peltonen, J.; Peltonen, S.; et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget 2017, 8, 45825–45836. [Google Scholar] [CrossRef]
- Chai, D.; Shan, H.; Wang, G.; Li, H.; Fang, L.; Song, J.; Zhang, Q.; Bai, J.; Zheng, J. AIM2 is a potential therapeutic target in human renal carcinoma and suppresses its invasion and metastasis via enhancing autophagy induction. Exp. Cell Res. 2018, 370, 561–570. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Yu, S. AIM2 regulates viability and apoptosis in human colorectal cancer cells via the PI3K/Akt pathway. OncoTargets Ther. 2017, 10, 811–817. [Google Scholar] [CrossRef]
- Ma, X.; Guo, P.; Qiu, Y.; Mu, K.; Zhu, L.; Zhao, W.; Li, T.; Han, L. Loss of AIM2 expression promotes hepatocarcinoma progression through activation of mTOR-S6K1 pathway. Oncotarget 2016, 7, 36185–36197. [Google Scholar] [CrossRef]
- Chen, S.L.; Liu, L.L.; Lu, S.X.; Luo, R.Z.; Wang, C.H.; Wang, H.; Cai, S.H.; Yang, X.; Xie, D.; Zhang, C.Z.; et al. HBx-mediated decrease of AIM2 contributes to hepatocellular carcinoma metastasis. Mol. Oncol. 2017, 11, 1225–1240. [Google Scholar] [CrossRef]
- Chai, D.; Liu, N.; Li, H.; Wang, G.; Song, J.; Fang, L.; Lu, Z.; Yao, H.; Zheng, J. H1/pAIM2 nanoparticles exert anti-tumour effects that is associated with the inflammasome activation in renal carcinoma. J. Cell. Mol. Med. 2018, 22, 5670–5681. [Google Scholar] [CrossRef]
- Deswaerte, V.; Nguyen, P.; West, A.; Browning, A.F.; Yu, L.; Ruwanpura, S.M.; Balic, J.; Livis, T.; Girard, C.; Preaudet, A.; et al. Inflammasome Adaptor ASC Suppresses Apoptosis of Gastric Cancer Cells by an IL18-Mediated Inflammation-Independent Mechanism. Cancer Res. 2018, 78, 1293–1307. [Google Scholar] [CrossRef]
- Ahmed, S.G.; Ahmed, A.; Maguire, C.A.; Doha, M.; Sagers, J.; Lewis, R.M.; Muzikansky, A.; Giovannini, M.; Stemmer-Rachamimov, A.; Stankovic, K.M.; et al. Gene therapy with apoptosis-associated speck-like protein (ASC), a newly described schwannoma tumor suppressor, inhibits schwannoma growth in vivo. Neuro Oncol. 2019. [Google Scholar] [CrossRef]
- Kitazawa, M.; Hida, S.; Fujii, C.; Taniguchi, S.; Ito, K.; Matsumura, T.; Okada, N.; Sakaizawa, T.; Kobayashi, A.; Takeoka, M.; et al. ASC Induces Apoptosis via Activation of Caspase-9 by Enhancing Gap Junction-Mediated Intercellular Communication. PLoS ONE 2017, 12, e0169340. [Google Scholar] [CrossRef]
- Sakaizawa, T.; Matsumura, T.; Fujii, C.; Hida, S.; Toishi, M.; Shiina, T.; Yoshida, K.; Hamanaka, K.; Ito, K.I.; Taniguchi, S. Potential Role of ASC, a Proapoptotic Protein, for Determining the Cisplatin Susceptibility of Lung Cancer Cells. Tohoku J. Exp. Med. 2018, 244, 133–144. [Google Scholar] [CrossRef]
- Graf, R.P.; Keller, N.; Barbero, S.; Stupack, D. Caspase-8 as a regulator of tumor cell motility. Curr. Mol. Med. 2014, 14, 246–254. [Google Scholar] [CrossRef]
- Okada, N.; Fujii, C.; Matsumura, T.; Kitazawa, M.; Okuyama, R.; Taniguchi, S.; Hida, S. Novel role of ASC as a regulator of metastatic phenotype. Cancer Med. 2016, 5, 2487–2500. [Google Scholar] [CrossRef]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Wu, C.S.; Chang, K.P.; OuYang, C.N.; Kao, H.K.; Hsueh, C.; Chen, L.C.; Cheng, H.Y.; Liang, Y.; Liou, W.; Liang, C.L.; et al. ASC contributes to metastasis of oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 50074–50085. [Google Scholar] [CrossRef]
- Baldwin, A.G.; Brough, D.; Freeman, S. Inhibiting the Inflammasome: A Chemical Perspective. J. Med. Chem. 2016, 59, 1691–1710. [Google Scholar] [CrossRef]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; Di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Thi, H.T.H.; Hong, S. Inflammasome as a Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 62–73. [Google Scholar] [CrossRef]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Subramaniyam, N.; Arumugam, S.; Ezthupurakkal, P.B.; Ariraman, S.; Biswas, I.; Muthuvel, S.K.; Balakrishnan, A.; Alshammari, G.M.; Chinnasamy, T. Unveiling anticancer potential of glibenclamide: Its synergistic cytotoxicity with doxorubicin on cancer cells. J. Pharm. Biomed. Anal. 2018, 154, 294–301. [Google Scholar] [CrossRef]
- Yan, B.; Peng, Z.; Xing, X.; Du, C. Glibenclamide induces apoptosis by activating reactive oxygen species dependent JNK pathway in hepatocellular carcinoma cells. Biosci. Rep. 2017, 37, BSR20170685. [Google Scholar] [CrossRef]
- Yasukagawa, T.; Niwa, Y.; Simizu, S.; Umezawa, K. Suppression of cellular invasion by glybenclamide through inhibited secretion of platelet-derived growth factor in ovarian clear cell carcinoma ES-2 cells. FEBS Lett. 2012, 586, 1504–1509. [Google Scholar] [CrossRef]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.B.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, Q.; Hu, N.; Zheng, F.; Zhang, X.; Ni, Y.; Liu, J. Downregulation of hsa_circ_0068087 ameliorates TLR4/NF-kappaB/NLRP3 inflammasome-mediated inflammation and endothelial cell dysfunction in high glucose conditioned by sponging miR-197. Gene 2019, 709, 1–7. [Google Scholar] [CrossRef]
- Song, J.; Yang, S.; Yin, R.; Xiao, Q.; Ma, A.; Pan, X. MicroRNA-181a regulates the activation of the NLRP3 inflammatory pathway by targeting MEK1 in THP-1 macrophages stimulated by ox-LDL. J. Cell. Biochem. 2019, 120, 13640–13650. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, S.; Wu, J.; Chen, M.; Cai, M.C.; Fu, Y.; Li, W.; Wang, J.; Zhao, X.; Yu, Z.; et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin. Cancer Res. 2018, 24, 6066–6077. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.; Kelliher, M.A.; et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef]
- Ratner, D.; Orning, M.P.; Proulx, M.K.; Wang, D.; Gavrilin, M.A.; Wewers, M.D.; Alnemri, E.S.; Johnson, P.F.; Lee, B.; Mecsas, J.; et al. The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. PLoS Pathog. 2016, 12, e1006035. [Google Scholar] [CrossRef]
- Ratner, D.; Orning, M.P.; Lien, E. Bacterial secretion systems and regulation of inflammasome activation. J. Leukoc. Biol. 2017, 101, 165–181. [Google Scholar] [CrossRef]
- Lee, S.; Hirohama, M.; Noguchi, M.; Nagata, K.; Kawaguchi, A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Cookson, B.T. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007, 3, e161. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Gao, J.; Qiu, X.; Xi, G.; Liu, H.; Zhang, F.; Lv, T.; Song, Y. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 2018, 40, 1971–1984. [Google Scholar] [CrossRef]
- Wang, W.J.; Chen, D.; Jiang, M.Z.; Xu, B.; Li, X.W.; Chu, Y.; Zhang, Y.J.; Mao, R.; Liang, J.; Fan, D.M. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis. 2018, 19, 74–83. [Google Scholar] [CrossRef]
- Ahmed, S.G.; Abdelanabi, A.; Doha, M.; Brenner, G.J. Schwannoma gene therapy by adeno-associated virus delivery of the pore-forming protein Gasdermin-D. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef]
- Lage, S.L.; Dominical, V.M.; Wong, C.S.; Sereti, I. Evaluation of Canonical Inflammasome Activation in Human Monocytes by Imaging Flow Cytometry. Front. Immunol. 2019, 10, 1284. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. CB 2016, 26, R568–R572. [Google Scholar] [CrossRef]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Munoz-Wolf, N.; Lavelle, E.C. A Guide to IL-1 family cytokines in adjuvanticity. FEBS J. 2018, 285, 2377–2401. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Wu, J.; Hong, Y.; Wu, T.; Wang, J.; Chen, X.; Wang, Z.; Cheng, B.; Xia, J. Stromal-epithelial lactate shuttle induced by tumorderived interleukin1beta promotes cell proliferation in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 41, 687–696. [Google Scholar] [CrossRef]
- Hu, M.; Yuan, X.; Liu, Y.; Tang, S.; Miao, J.; Zhou, Q.; Chen, S. IL-1beta-induced NF-kappaB activation down-regulates miR-506 expression to promotes osteosarcoma cell growth through JAG1. Biomed. Pharmacother. 2017, 95, 1147–1155. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, Y.; Chen, X.; Peng, D. IL-1beta-mediated NF-kappaB signaling augments the osteosarcoma cell growth through modulating miR-376c/TGFA axis. Die Pharm. 2017, 72, 419–424. [Google Scholar] [CrossRef]
- Yu, A.; Wang, Y.; Bian, Y.; Chen, L.; Guo, J.; Shen, W.; Chen, D.; Liu, S.; Sun, X. IL-1beta promotes the nuclear translocaiton of S100A4 protein in gastric cancer cells MGC803 and the cell’s stem-like properties through PI3K pathway. J. Cell. Biochem. 2018, 119, 8163–8173. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Xie, J.; Hou, D.; Zhang, H.; Huang, H. Melatonin inhibits epithelialtomesenchymal transition in gastric cancer cells via attenuation of IL1beta/NFkappaB/MMP2/MMP9 signaling. Int. J. Mol. Med. 2018, 42, 2221–2228. [Google Scholar] [CrossRef]
- Li, T.; Zhu, J.; Zuo, S.; Chen, S.; Ma, J.; Ma, Y.; Guo, S.; Wang, P.; Liu, Y. 1,25(OH)2D3 attenuates IL-1beta-induced epithelial to mesenchymal transition through inhibiting the expression of lncTCF7. Oncol. Res. 2018, 27, 739–750. [Google Scholar] [CrossRef]
- Castano, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. IL-1beta inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097. [Google Scholar] [CrossRef]
- Jimenez-Garduno, A.M.; Mendoza-Rodriguez, M.G.; Urrutia-Cabrera, D.; Dominguez-Robles, M.C.; Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Storr, S.J.; Safuan, S.; Ahmad, N.; El-Refaee, M.; Jackson, A.M.; Martin, S.G. Macrophage-derived interleukin-1beta promotes human breast cancer cell migration and lymphatic adhesion in vitro. Cancer Immunol. Immunother. CII 2017, 66, 1287–1294. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1beta activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, S.; Liu, T.; Liu, Q.; Chen, Y.; Tan, D.; Ma, R.; Lu, X. IL-1beta from M2 macrophages promotes migration and invasion of ESCC cells enhancing epithelial-mesenchymal transition and activating NF-kappaB signaling pathway. J. Cell. Biochem. 2018, 119, 7040–7052. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef]
- Wang, C.I.; Yu, C.J.; Huang, Y.; Yi, J.S.; Cheng, H.W.; Kao, H.K.; Lao, W.W.; Chang, K.P. Association of overexpressed karyopherin alpha 2 with poor survival and its contribution to interleukin-1beta-induced matrix metalloproteinase expression in oral cancer. Head Neck 2018, 40, 1719–1733. [Google Scholar] [CrossRef]
- Lee, M.K.; Park, J.H.; Gi, S.H.; Hwang, Y.S. IL-1beta Induces Fascin Expression and Increases Cancer Invasion. Anticancer Res. 2018, 38, 6127–6132. [Google Scholar] [CrossRef]
- Chen, X.; Lv, Q.; Hong, Y.; Chen, X.; Cheng, B.; Wu, T. IL-1beta maintains the redox balance by regulating glutaredoxin 1 expression during oral carcinogenesis. J. Oral Pathol. Med. 2017, 46, 332–339. [Google Scholar] [CrossRef]
- Mendoza-Rodriguez, M.G.; Ayala-Sumuano, J.T.; Garcia-Morales, L.; Zamudio-Meza, H.; Perez-Yepez, E.A.; Meza, I. IL-1beta Inflammatory Cytokine-Induced TP63 Isoform NP63alpha Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 270. [Google Scholar] [CrossRef]
- Mendoza-Rodriguez, M.; Arevalo Romero, H.; Fuentes-Panana, E.M.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef]
- Dinarello, C.A. An expanding role for interleukin-1 blockade from gout to cancer. Mol. Med. 2014, 20 (Suppl. 1), S43–S58. [Google Scholar] [CrossRef]
- Braddock, M.; Quinn, A. Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004, 3, 330–339. [Google Scholar] [CrossRef]
- Fleischmann, R.; Stern, R.; Iqbal, I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin. Biol. Ther. 2004, 4, 1333–1344. [Google Scholar] [CrossRef]
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jorgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 2016, 128, 2671–2682. [Google Scholar] [CrossRef]
- Kurzrock, R.; Hickish, T.; Wyrwicz, L.; Saunders, M.; Wu, Q.; Stecher, M.; Mohanty, P.; Dinarello, C.A.; Simard, J. Interleukin-1 receptor antagonist levels predict favorable outcome after bermekimab, a first-in-class true human interleukin-1alpha antibody, in a phase III randomized study of advanced colorectal cancer. Oncoimmunology 2019, 8, 1551651. [Google Scholar] [CrossRef]
- Stanam, A.; Gibson-Corley, K.N.; Love-Homan, L.; Ihejirika, N.; Simons, A.L. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 76087–76100. [Google Scholar] [CrossRef]
- Isambert, N.; Hervieu, A.; Rebe, C.; Hennequin, A.; Borg, C.; Zanetta, S.; Chevriaux, A.; Richard, C.; Derangere, V.; Limagne, E.; et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): A single-arm phase 2 study. Oncoimmunology 2018, 7, e1474319. [Google Scholar] [CrossRef]
- Zhuang, Z.; Ju, H.Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-kappaB Activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef]
- Church, L.D.; McDermott, M.F. Canakinumab, a fully-human mAb against IL-1beta for the potential treatment of inflammatory disorders. Curr. Opin. Mol. Ther. 2009, 11, 81–89. [Google Scholar]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef]
- Orlando, I.; Vitale, A.; Rigante, D.; Lopalco, G.; Fabiani, C.; Cantarini, L. Long-term efficacy and safety of the interleukin-1 inhibitors anakinra and canakinumab in refractory Behcet disease uveitis and concomitant bladder papillary carcinoma. Intern. Med. J. 2017, 47, 1086–1088. [Google Scholar] [CrossRef]
- Thompson, P.L.; Nidorf, S.M. Anti-inflammatory therapy with canakinumab for atherosclerotic disease: Lessons from the CANTOS trial. J. Thorac. Dis. 2018, 10, 695–698. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef]
- Garg, M.; de Jesus, A.A.; Chapelle, D.; Dancey, P.; Herzog, R.; Rivas-Chacon, R.; Muskardin, T.L.W.; Reed, A.; Reynolds, J.C.; Goldbach-Mansky, R.; et al. Rilonacept maintains long-term inflammatory remission in patients with deficiency of the IL-1 receptor antagonist. JCI Insight 2017, 2, e94838. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef]
- Wawrocki, S.; Druszczynska, M.; Kowalewicz-Kulbat, M.; Rudnicka, W. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim. Pol. 2016, 63, 59–63. [Google Scholar] [CrossRef]
- Enoksson, S.L.; Grasset, E.K.; Hagglof, T.; Mattsson, N.; Kaiser, Y.; Gabrielsson, S.; McGaha, T.L.; Scheynius, A.; Karlsson, M.C. The inflammatory cytokine IL-18 induces self-reactive innate antibody responses regulated by natural killer T cells. Proc. Natl. Acad. Sci. USA 2011, 108, E1399–E1407. [Google Scholar] [CrossRef]
- Sedimbi, S.K.; Hagglof, T.; Karlsson, M.C. IL-18 in inflammatory and autoimmune disease. Cell. Mol. Life Sci. CMLS 2013, 70, 4795–4808. [Google Scholar] [CrossRef]
- Jurecekova, J.; Babusikova, E.; Kmetova Sivonova, M.; Drobkova, H.; Petras, M.; Kliment, J.; Halasova, E. Association between interleukin-18 variants and prostate cancer in Slovak population. Neoplasma 2017, 64, 148–155. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chang, W.S.; Tsai, C.W.; Hsia, T.C.; Shen, T.C.; Bau, D.T.; Shui, H.A. Interleukin-18 promoter genotype is associated with the risk of nasopharyngeal carcinoma in Taiwan. Cancer Manag. Res. 2018, 10, 5199–5207. [Google Scholar] [CrossRef]
- Bakr, N.M.; Awad, A.; Moustafa, E.A. Association of genetic variants in the interleukin-18 gene promoter with risk of hepatocellular carcinoma and metastasis in patients with hepatitis C virus infection. IUBMB Life 2018, 70, 165–174. [Google Scholar] [CrossRef]
- Gan, W.Y.; Li, H.M.; Zhang, Y.G.; Li, C.M.; Wang, Y. Association between IL18-607C/A and -137G/C polymorphisms and susceptibility to non-small cell lung cancer in a Chinese population. Genet. Mol. Res. GMR 2016, 15, gmr15048822. [Google Scholar] [CrossRef]
- Wang, H.; Hua, M.; Wang, S.; Yu, J.; Chen, C.; Zhao, X.; Zhang, C.; Zhong, C.; Wang, R.; He, N.; et al. Genetic polymorphisms of IL-18 rs1946518 and IL-1beta rs16944 are associated with prognosis and survival of acute myeloid leukemia. Inflamm. Res. 2017, 66, 249–258. [Google Scholar] [CrossRef]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chretien, M.L.; Guillerey, C.; Putz, E.M.; Bald, T.; Forster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648.e5. [Google Scholar] [CrossRef]
- Ko, C.Y.; Wang, W.L.; Li, C.F.; Jeng, Y.M.; Chu, Y.Y.; Wang, H.Y.; Tseng, J.T.; Wang, J.M. IL-18-induced interaction between IMP3 and HuR contributes to COX-2 mRNA stabilization in acute myeloid leukemia. J. Leukoc. Biol. 2016, 99, 131–141. [Google Scholar] [CrossRef]
- Ding, S.; Tang, Z.; Jiang, Y.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. IL-8 Is Involved in Estrogen-Related Receptor alpha-Regulated Proliferation and Migration of Colorectal Cancer Cells. Dig. Dis. Sci. 2017, 62, 3438–3446. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Z.; Li, J.; Ban, S.; Duan, C.; Liu, W. Interleukin-18 expression in oral squamous cell carcinoma: its role in tumor cell migration and invasion, and growth of tumor cell xenografts. FEBS Open Bio 2018, 8, 1953–1963. [Google Scholar] [CrossRef]
- Usul Afsar, C.; Karabulut, M.; Karabulut, S.; Alis, H.; Gonenc, M.; Dagoglu, N.; Serilmez, M.; Tas, F. Circulating interleukin-18 (IL-18) is a predictor of response to gemcitabine based chemotherapy in patients with pancreatic adenocarcinoma. J. Infect. Chemother. 2017, 23, 196–200. [Google Scholar] [CrossRef]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675. [Google Scholar] [CrossRef]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef]
- Li, J.; Qiu, G.; Fang, B.; Dai, X.; Cai, J. Deficiency of IL-18 Aggravates Esophageal Carcinoma Through Inhibiting IFN-gamma Production by CD8(+)T Cells and NK Cells. Inflammation 2018, 41, 667–676. [Google Scholar] [CrossRef]
- Xu, X.; Song, C.; Chen, Z.; Yu, C.; Wang, Y.; Tang, Y.; Luo, J. Downregulation of HuR Inhibits the Progression of Esophageal Cancer through Interleukin-18. Cancer Res. Treat. 2018, 50, 71–87. [Google Scholar] [CrossRef]
- Senju, H.; Kumagai, A.; Nakamura, Y.; Yamaguchi, H.; Nakatomi, K.; Fukami, S.; Shiraishi, K.; Harada, Y.; Nakamura, M.; Okamura, H.; et al. Effect of IL-18 on the Expansion and Phenotype of Human Natural Killer Cells: Application to Cancer Immunotherapy. Int. J. Biol. Sci. 2018, 14, 331–340. [Google Scholar] [CrossRef]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842. [Google Scholar] [CrossRef]
- Yang, C.; Cao, H.; Liu, N.; Xu, K.; Ding, M.; Mao, L.J. Oncolytic adenovirus expressing interleukin-18 improves antitumor activity of dacarbazine for malignant melanoma. Drug Des. Dev. Ther. 2016, 10, 3755–3761. [Google Scholar] [CrossRef]
- Markowitz, G.J.; Yang, P.; Fu, J.; Michelotti, G.A.; Chen, R.; Sui, J.; Yang, B.; Qin, W.H.; Zhang, Z.; Wang, F.S.; et al. Inflammation-Dependent IL18 Signaling Restricts Hepatocellular Carcinoma Growth by Enhancing the Accumulation and Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 2016, 76, 2394–2405. [Google Scholar] [CrossRef]
- Zhuang, L.; Fulton, R.J.; Rettman, P.; Sayan, A.E.; Coad, J.; Al-Shamkhani, A.; Khakoo, S.I. Activity of IL-12/15/18 primed natural killer cells against hepatocellular carcinoma. Hepatol. Int. 2019, 13, 75–83. [Google Scholar] [CrossRef]
- Liu, X.; Hu, J.; Li, Y.; Cao, W.; Wang, Y.; Ma, Z.; Li, F. Mesenchymal stem cells expressing interleukin-18 inhibit breast cancer in a mouse model. Oncol. Lett. 2018, 15, 6265–6274. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, M.; Feng, Y.; He, R.; Xu, X.; Xie, Y.; Shi, X.; Zhou, M.; Pan, S.; Wang, M.; et al. Regulatory B cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway. Oncotarget 2018, 9, 14803–14814. [Google Scholar] [CrossRef]
- Guo, X.; Zheng, L.; Jiang, J.; Zhao, Y.; Wang, X.; Shen, M.; Zhu, F.; Tian, R.; Shi, C.; Xu, M.; et al. Blocking NF-kappaB Is Essential for the Immunotherapeutic Effect of Recombinant IL18 in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 5939–5950. [Google Scholar] [CrossRef]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef]
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980. [Google Scholar] [CrossRef]
- Robertson, M.J.; Kirkwood, J.M.; Logan, T.F.; Koch, K.M.; Kathman, S.; Kirby, L.C.; Bell, W.N.; Thurmond, L.M.; Weisenbach, J.; Dar, M.M. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin. Cancer Res. 2008, 14, 3462–3469. [Google Scholar] [CrossRef]
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Millward, M.; Mainwaring, P.; Kefford, R.; Logan, T.; Pavlick, A.; Kathman, S.J.; Laubscher, K.H.; Dar, M.M.; Kirkwood, J.M. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer 2009, 115, 859–868. [Google Scholar] [CrossRef]
- Robertson, M.J.; Kline, J.; Struemper, H.; Koch, K.M.; Bauman, J.W.; Gardner, O.S.; Murray, S.C.; Germaschewski, F.; Weisenbach, J.; Jonak, Z.; et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J. Immunother. 2013, 36, 331–341. [Google Scholar] [CrossRef]
- Simpkins, F.; Flores, A.; Chu, C.; Berek, J.S.; Lucci, J., 3rd; Murray, S.; Bauman, J.; Struemper, H.; Germaschewski, F.; Jonak, Z.; et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer Immunol. Res. 2013, 1, 168–178. [Google Scholar] [CrossRef]
- Cao, Q.; Cai, W.; Niu, G.; He, L.; Chen, X. Multimodality imaging of IL-18–binding protein-Fc therapy of experimental lung metastasis. Clin. Cancer Res. 2008, 14, 6137–6145. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313. https://doi.org/10.3390/cancers11091313
Wang M, Jiang S, Zhang Y, Li P, Wang K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers. 2019; 11(9):1313. https://doi.org/10.3390/cancers11091313
Chicago/Turabian StyleWang, Man, Shuai Jiang, Yinfeng Zhang, Peifeng Li, and Kun Wang. 2019. "The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer" Cancers 11, no. 9: 1313. https://doi.org/10.3390/cancers11091313
APA StyleWang, M., Jiang, S., Zhang, Y., Li, P., & Wang, K. (2019). The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers, 11(9), 1313. https://doi.org/10.3390/cancers11091313