Design of Outer Membrane Vesicles as Cancer Vaccines: A New Toolkit for Cancer Therapy


Abstract
1. Introduction
2. Vaccines in Cancer Therapy
2.1. Historical Overview
2.2. Status of Clinical Trials
3. New OMV Applications: Tumor Vaccines
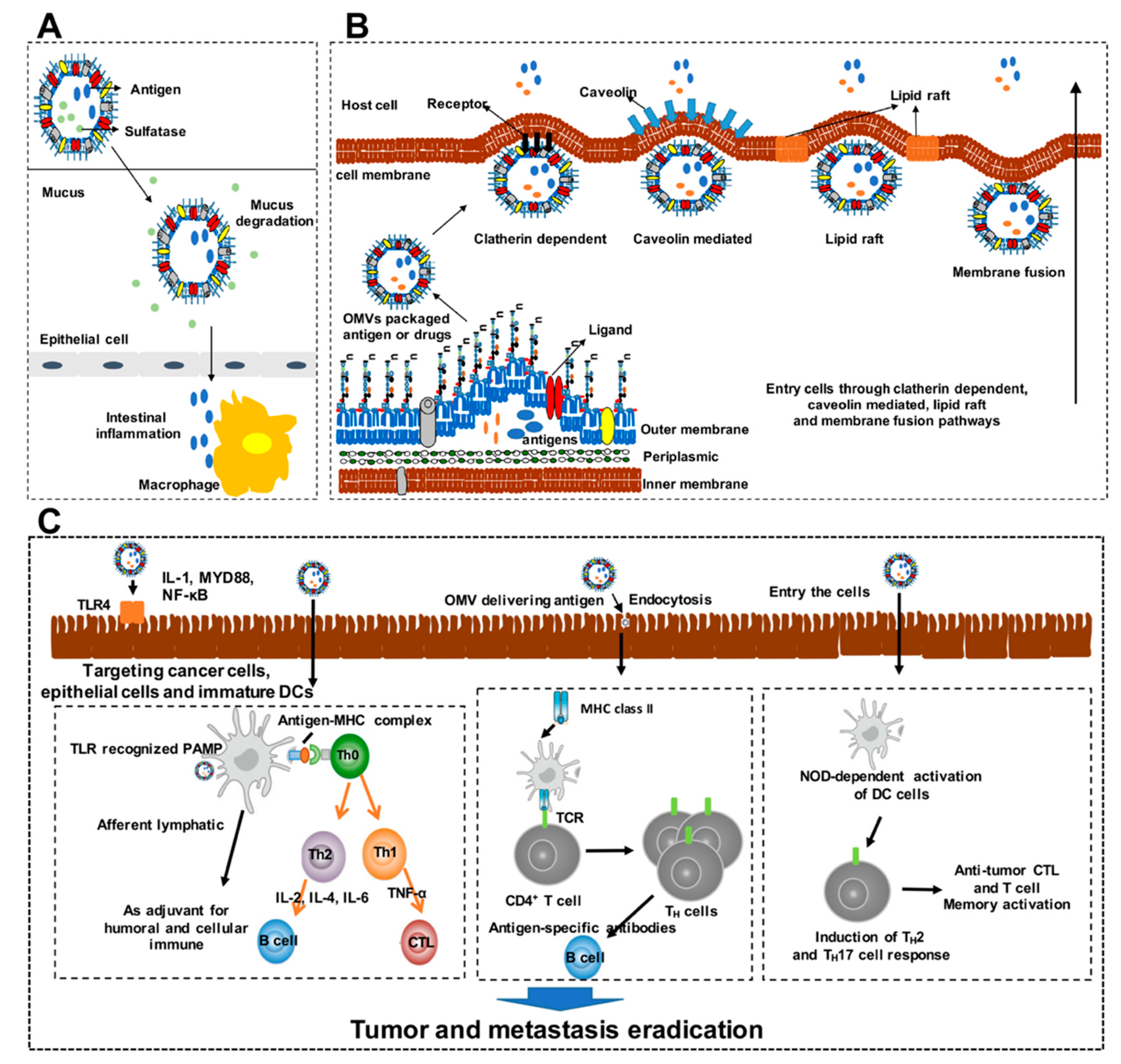
3.1. Roles of OMVs as Vaccines

{kind=link}
{kind=link}
| OMV Source | Model Establishment | Animal Model | Route of Administration | Adjuvant Used | Protection | Resulting Immune Response | References |
|---|---|---|---|---|---|---|---|
| A. baumannii | Sepsis | Female ICR Mice (6–8 weeks of age) | Intramuscular | Alum | 73% survival (compared with 7% in controls) | Serum IgG | [54] |
| Acute pneumonia | Reduced bacterial burden in the lungs, spleen and BALF | BALF IgA and IgG | |||||
| B. burgdorferi | Infection | New Zealand White Rabbits | Intramuscular | Alum | Reduced bacterial burden in the skins | Serum IgG | [55] |
| B. pseudomallei | Septicemic infection | Female BALB/c Mice (8–10 weeks of age) | Subcutaneous | None | 50% survival (compared with 0% in controls) | Serum IgG and TH1 cell response | [56] |
| B. pertussis | Infection | Female C57BL/6 Mice (8 weeks of age) | Intraperitoneal | Alum | Reduced bacterial burden in the lungs | Serum IgG, TH1, and TH17 response | [57] |
| E. coli | Sepsis | Both C57BL/6 and BALB/c Mice (5 weeks of age) | Intraperitoneal | None | 80%–100% survival (compared with 20% in controls) | Serum IgG, TH1, and TH17 response | [58] |
| E. coli expressing streptococcal antigen | Sepsis (group A streptococci) | CD1 female Mice | Intraperitoneal | Alum | 100% survival (compared with 20% in controls) | Serum IgG1 and IgG2a | [19] |
| E. coli expressing Apx fusion antigen | Porcine pleuropneumonia | Female BALB/c Mice (4–5 weeks of age) | Subcutaneous | Alum | 62.5% and 87.5% survival for Actinobacillus pleuropneumoniae infection | Serum IgG TH1 and TH2 cytokines secretion | [59] |
| Nontypeable H. influenzae | Infection | Female BALB/c Mice | Intranasal | None | Reduced bacterial burden in the nasopharynx | Serum IgA, IgG1, and IgM | [20] |
| H. pylori | Infection | Female BALB/c Mice | Intragastric | Cholera toxin | 100% protection (compared with 20% in controls) | Serum IgG | [60] |
| K. pneumoniae | Sepsis | Female C57BL/6 Mice (6–7 weeks of age) | Intraperitoneal | None | 80%–100% survival (compared with control groups) | Serum IgG and the secretion of key cytokines of TH1 cells | [61] |
| N. meningitides* | Meningococcal infection | Human use | Intramuscular | Alum | Provided broad-based protection | Not mentioned | [62] |
| N. meningitides with inactivated RSV | Infection (RSV) | Female BALB/c Mice (5–8 weeks of age) | Intranasal | OMVs | 100% protection as measured by viral load | IgA plasma cells in NALT, IgA, IgG1, IgG2a, and IgG2b in lung and serum | [63] |
| P. gingivalis | Infection | Female BALB/c Mice (6–8 weeks of age) | Intranasal | Poly (I:C) | Not mentioned | Serum IgG (including IgG1 and IgG2a), salivary S-IgA, and cytokine secretion | [64] |
| S. enteritidis | Foodborne infections | Female BALB/c Mice (6–8 weeks of age) | Intranasal or intraperitoneal | None | 83.3%–91% survival (compared with 0% in controls) | Serum IgG and secretory IgA | [65] |
| S. typhimurium | Infection | Female C3H/HeJ and C3H/HeN Mice (6–8 weeks of age) | Intraperitoneal | None | Reduced bacterial burden in spleen, liver, MLNs, and Peyer’s patches | Serum IgG and IFNγ-producing CD4+ T cells | [66] |
| S. flexneri | Infection (lethal dose) | Female BALB/c Mice (9 weeks of age) | Nasal or oral | Poly-anhydride nanoparticles | 80%–100% protection (compared with 0% in controls) | Serum IgG1 and IgG2a, fecal IgA | [67] |
| S. boydii | Infection (lethal dose) | Female BALB/c Mice (6–7 weeks of age) | Oral | None | 100% protection (compared with 0% in controls) | Mucosal IgG and IgA, TH1 cell response | [68] |
| V. cholerae | Infection (neonates) | BALB/c Mice (5- to 6-day-old pups) | Intragastric or intranasal | None | Significantly reduced colonization of neonates | Serum IgA, IgG1, IgG2a, and IgM | [69] |
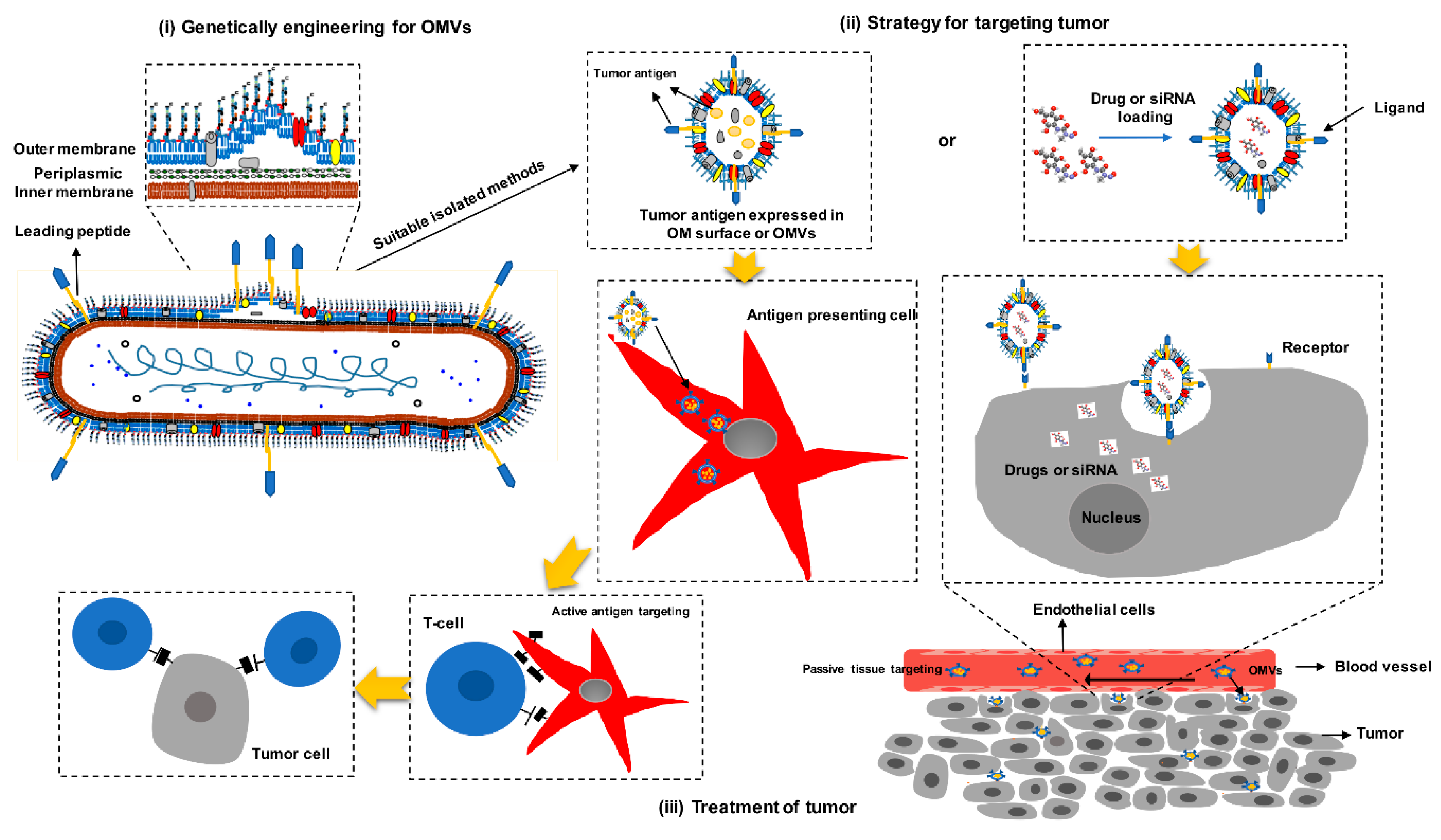
3.2. Mechanisms of OMVs Design as Cancer Vaccines
3.3. Advantages of OMVs as Cancer Vaccines
3.4. Bacterial OMVs as Cancer Vaccines
| Vaccine Strain | Gene Mutated or Modified | Descriptions | Therapeutic Agents (Tumor Antigen, Immune molecule, Anti-Tumor Drug) | Prokaryotic/ Eukaryotic Expression | Cancer Indication | Clinical Trials | Therapeutic Efficacy# | References |
|---|---|---|---|---|---|---|---|---|
| Salmonella | ||||||||
| VNP20009 | msbB/Pur | The deletion of msbB modifies the lipid-A structure reducing bacterial ability of TNF-α induction and mutation of pur results in bacterial deficiency in adenine synthesis | Cytokines: IL-18, LIGHT, CCL21; Cytotoxic agents: TRAIL, FasL; Regulators: Thrombospondin; TAA: CEA-scFv, TGF-α; Prodrug enzymes: Cytosine deaminase; si-RNA: sox2-specific, IDO-specific | Prokaryotic or eukaryotic expression | Colon, lung, breast, cervical melanoma | Phase I (used for metastatic melanoma) | Safety, and targeting to tumor cells, but no patients experienced objective tumor regression in Phase I clinical trial | [113,114,115,116,117,118,119,120,121,122] |
| SL3261 SL7207 et. al. | aro- | The genes aroA and aroD are responsible for the biosynthesis of aromatic amino acids | Cytokines: IL-2, IL-12, IL-4, IL-18, IFN-γ, GM-CSF; Cytotoxic agents: cytolysin A, Noxa, FlaB, apoptin, diphtheria toxin; Regulators: 4-1BBL; TAA: CD20-specific antibody, HPV16-E7, Survivin, FLK-1; si-RNA: Bcl-2-specific | Prokaryotic or eukaryotic expression | Osteosarcoma, melanoma, colon, breast, cervical, gastric, neuroblastoma, lung, prostate | None | Effectively suppressed tumor growth and metastasis in mouse model | [123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139] |
| SHJ2037 | relA/spoT | Lacking both RelA and SpoT, cells are unable to produce ppGpp, a global regulator involving bacterial adaptation of extreme environment | Regulator: L-asparaginase, TAA: RGD peptides, TGF-α | Prokaryotic expression | Colon, breast | None | Effectively suppressed various solid tumor growth in mouse model | [140,141] |
| ST8 | asd/gmd | The gene gmd is in the colanic acid gene cluster and encodes GDP-mannose 4,6-dehydratase; Salmonella asd mutants will lyse during growth unless exogenous DAP is supplied | Regulator: Endostatin | Prokaryotic expression | Colon | None | Successfully suppressed angiogenesis and consequently retards tumor growth | [142] |
| LH430 | phoP/phoQ | The knock-out of PhoP and PhoQ that regulate acid phosphatase synthesis significantly reduces bacterial survival in macrophages | Regulators: Endostatin; siRNA: STAT3-specific | Eukaryotic expression | Hepatoma | None | Stimulated apoptosis and inhibited angiogenesis in tumors | [143] |
| MvP728 | purD/htrA | The gene purD encodes 5′- phosphoribosylglycinamide synthetase involved in purine biosynthesis; htrA encodes heat-shock proteins that are important for virulence of the bacterium | TAA: survivin | Eukaryotic expression | Glioblastoma, colon | None | Enhanced effector-memory CTL response and inhibited tumor growth in mouse model | [144] |
| χ4550 | cya/crp | The two genes cya and crp encode cAMP (cyclic adenosine monophosphate) synthetase and cAMP receptor protein | Cytokines: IL-2, TNF-α | Prokaryotic expression | Melanoma | None | Inhibited tumor growth as well as enhanced host survival | [145] |
| RE88 | dam | The gene dam encodes DNA, adenine methylase sptP is an effector protein of Salmonella | TAA: legumain | Eukaryotic expression | Breast | None | Effectively suppressed tumor angiogenesis | [146] |
| SB824 | sptP | SptP is an effector protein of Salmonella pathogenicity island 1 (SPI-1), that acts as protein tyrosine phosphatase/GTPase activating proteins | TAA: YopE1-138/p60130-477/M45 | Prokaryotic expression | Fibrosarcoma | None | Showed complete tumor regression | [147] |
| Listeria* | ||||||||
| ADXS11-001 ADXS31-142 ADXS31-164 ADXS-NEO et. al. | tLLO (Lysteriolysin) | Lm has the ability to replicate in the cytosol of APCs after escaping from the phagolysosome, which requires the virulence factor listeriolysin O (LLO) protein, and targeted antigen fused to a non-hemolytic LLO | TAA: HPV 16 E7, PSA, VEGFR2, HER2, Personal Neo-antigens | Prokaryotic expression | Cervical, oropharyngeal, prostate, colon, lung, breast, HER2+ solid tumors | Phase I (used for cervical cancer) | 36% survival for 12 months and 11% response rate were observed in patients | [148,149,150] |
| CRS-100 CRS-207 ADU-623 ADU-214 et. al. | actA/inlB | Two virulence genes, actA and internalin B (InlB), and their combined deletion results in 1000-fold attenuation when compared to wildtype | TAA: Mesothelin, EGFRvIII, NY-ESO-1, Personal Neo-antigens | Prokaryotic expression | Pancreatic, lung, ovarian, mesothelioma, prostate | Phase I/II (used for pancreatic cancer and mesothelioma | 37% of patients survived 15 months or more, and the combination with chemotherapy is more effective | [151,152] |
| Attenuated L. monocytogenes | dal/dat | In the absence of dal and dat expression, replication of LM can depend only on the availability of exogenous D-alanine. After introduction of the dal and dat genes from Bacillus subtilis, the strain was able to synthesize D-alanine and to replicate to a limited extent that did not cause severe organ injuries | TAA: CD24 | Eukaryotic expression | Hepatocellular carcinoma | None | Significantly reduced the tumor size in mice and increased their survival from 0% to 48% | [153] |
| Clostridium* | ||||||||
| C. beijerinckii | Non | E. coli nitroreductase known to activate the nontoxic prodrug CB 1954 to a toxic anticancer drug | Prodrug enzymes: CB1954 | Prokaryotic expression | Breast | None | Lack of toxicity and highly selective growth in tumors | [154] |
| C. acetobutylicum | Non | C. acetobutylicum was genetically engineered to express and secrete either mTNF-alpha, IL-2, or the E. coli cytosine deaminase | Cytokines: TNF-α, IL-2; Cytotoxic agents: cytosine deaminase | Prokaryotic expression | Rhabdomyosarcoma | None | Safety and selective colonization pattern | [155] |
| Bifidobacterium* | ||||||||
| B. longum | Non | Bifidobacterium can selectively germinate and grow in the hypoxic regions of solid tumors after intravenous injection | Cytotoxic agents: cytosine deaminase, TRAIL; Prodrug enzymes: 5-fluorocytosine; TAA: Wilms’ tumor 1 | Prokaryotic expression | Lung, melanoma; leukemia | None | Selectively proliferated in tumors and significantly suppressed tumor weight and tumor growth | [156,157,158,159] |
| B. adolescentis | Non | A shuttle vector, pBV220 was used for expressing antigens | Regulator: Endostatin | Prokaryotic expression | Liver | None | Tumor growth in mice was inhibited by 23.1% | [160] |
| B. infantis | Non | B. infantis can selectively localize and proliferate in the hypoxic environment in several types of solid tumors | TAA: sFlt-1 | Eukaryotic expression | Lung | None | Inhibit the tumor growth and prolong survival time from 41 days to 51 days | [161] |
| Pseudomonas aeruginosa | ||||||||
| CHA-OST et. al. | exoS/exoT/aroA/lasI, fusion with EXO-S | An attenuated live bacterial vector using the type III secretion system (TTSS) of Pseudomonas aeruginosa to deliver in vivo tumor antigens | TAA: TRP-2, gp100, MUC18 | Prokaryotic expression | Glioma | None | 100% protection in prophylactic antitumor assay and 37.5% protection in therapeutic antitumor assay | [162,163] |
| Escherichia coli | ||||||||
| χ6212 | asd, SAH was cloned into E. coli | Staphylococcus aureus α-hemolysin (SAH) is a pore-forming protein that is naturally secreted and kills mammalian cells | Cytotoxic agents:Staphylococcus aureus α-hemolysin (SAH) | Prokaryotic expression | Breast | None | Tumor volume was 59% of induction compared with control group | [164] |
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Bray, F. Global cancer statistics. CA Cancer J. Clin. 2018, 8, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Terabe, M.; Trepel, J.B.; Pastan, I.; Stroncek, D.F.; Morris, J.C.; Wood, L.V. Cancer vaccine strategies: Translation from mice to human clinical trials. Cancer Immunol. Immunother. 2018, 67, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.L.; Dos Santos, A.L.S.; Nunes, N.C.C.; de Moraes Lino da Silva, F.; Ferreira, C.G.M.; de Melo, A.C. Cancer immunotherapy: The art of targeting the tumor immune microenvironment. Cancer Chemother. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Kouro, T.; Igarashi, Y.; Kawahara, M.; Sasada, T. Prospects for a personalized peptide vaccine against lung cancer. Expert Rev. Vaccines 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Murphy, A.; Zheng, L. From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy. Adv. Cancer Res. 2019, 143, 63–144. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Ramamoorthi, G.; Jia, Y.; Faughn, J.; Wiener, D.; Awshah, S.; Kodumudi, K.; Czerniecki, B.J. Immunotherapy in breast cancer: Current status and future directions. Adv. Cancer Res. 2019, 143, 295–349. [Google Scholar] [CrossRef] [PubMed]
- Limacher, J.M.; Quoix, E. TG4010: A therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology 2012, 1, 791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Randeep, S.; Charles, B. L-BLP25: A peptide vaccine strategy in non small cell lung cancer. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2007, 13, 4652–4654. [Google Scholar]
- Rodríguez, P.C.; Rodríguez, G.; González, G.; Lage, A. Clinical development and perspectives of CIMAvax EGF, Cuban vaccine for non-small-cell lung cancer therapy. Med. Rev. 2010, 12, 17–23. [Google Scholar]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 2015, 7, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Knox, K.W.; Vesk, M.; Work, E. Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J. Bacteriol. 1966, 92, 1206–1217. [Google Scholar] [PubMed]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Shockman, G.D.; Barrett, J.F. Structure, function, and assembly of cell walls of gram-positive bacteria. Annu. Rev. Microbiol. 1983, 37, 501–527. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Kesty, N.C.; Kuehn, M.J. Incorporation of heterologous outer membrane and periplasmic proteins into Escherichia coli outer membrane vesicles. J. Biol. Chem. 2004, 279, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Fantappiè, L.; Santis, M.D.; Chiarot, E.; Carboni, F.; Bensi, G.; Jousson, O.; Margarit, I.; Grandi, G. Antibody-mediated immunity induced by engineered Escherichia coli OMVs carrying heterologous antigens in their lumen. J. Extracell. Vesicles 2014, 3, 24015. [Google Scholar] [CrossRef] [PubMed]
- Sandro, R.; Leitner, D.R.; Jeremy, I.; Kristina, S.P.; Feldman, M.F.; Georg, K.; Joachim, R.; Stefan, S. Intranasal immunization with nontypeable Haemophilus influenzae outer membrane vesicles induces cross-protective immunity in mice. PLoS ONE 2012, 7, 520–521. [Google Scholar]
- Wong, K.K.; Li, W.A.; Mooney, D.J.; Dranoff, G. Advances in Therapeutic Cancer Vaccines. Adv. Immunol. 2016, 130, 191–249. [Google Scholar] [CrossRef]
- Gillison, M.L.; Chaturvedi, A.K.; Lowy, D.R. HPV prophylactic vaccines and the potential prevention of noncervical cancers in both men and women. Cancer 2010, 113, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.E. The rationale for prophylactic cancer vaccines and need for a paradigm shift. Cancer Gene Ther. 2006, 13, 725. [Google Scholar] [CrossRef] [PubMed]
- Alex, K.; Hanna, M.G. Overview of the cancer vaccine field: Are we moving forward? Hum. Vaccines 2012, 8, 1135–1140. [Google Scholar]
- Zhang, X.; Sharma, P.K.; Peter Goedegebuure, S.; Gillanders, W.E. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine 2017, 35, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.J.; Guillerey, C.; Radford, K.J. Can Dendritic Cell Vaccination Prevent Leukemia Relapse? Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, K. Immunotherapy: Cancer vaccines on the move. Nat. Rev. Clin. Oncol. 2017, 15, 9. [Google Scholar] [CrossRef]
- Tran, T.; Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E. Therapeutic cancer vaccine: Building the future from lessons of the past. Semin. Immunopathol. 2019, 41, 69–85. [Google Scholar] [CrossRef]
- Collichio, F.A.; Amatruda, T.; Senzer, N.N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Doleman, S.; Ye, Y.; Vanderwalde, A.M. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J. Clin. Oncol. 2014, 9, 74–75. [Google Scholar]
- Thara, E.; Dorff, T.B.; Averia-Suboc, M.; Luther, M.; Reed, M.E.; Pinski, J.K.; Quinn, D.I. Immune response to sipuleucel-T in prostate cancer. Cancers 2012, 4, 420–441. [Google Scholar] [CrossRef]
- Cecco, S.; Muraro, E.; Giacomin, E.; Martorelli, D.; Lazzarini, R.; Baldo, P.; Dolcetti, R. Cancer vaccines in phase II/III clinical trials: State of the art and future perspectives. Curr. Cancer Drug Targets 2011, 11, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Gordon, L.; Wong, A.J. Targeting EGF receptor variant III: Tumor-specific peptide vaccination for malignant gliomas. Expert Rev. Vaccines 2012, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Elsamadicy, A.A.; Chongsathidkiet, P.; Desai, R.; Woroniecka, K.; Farber, S.H.; Fecci, P.E.; Sampson, J.H. Prospect of rindopepimut in the treatment of glioblastoma. Expert Opin. Biol. Ther. 2017, 17, 507. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Clive, K.S.; Ritesh, P.; Benavides, L.C.; Gates, J.D.; Sears, A.K.; Alexander, S.; Sathibalan, P. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients: From US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer 2012, 118, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Acquavella, N.; Yu, Z.; Restifo, N.P. Therapeutic cancer vaccines: Are we there yet? Immunol. Rev. 2011, 239, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; I Peter, S.; Taylor, E.J.M.; Lidia, R.; Bartosz, C.; Marko, S.; Gina, H.; Voicu, C. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; Rosenberg, S.A. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Burg, S.H.V.D.; Arens, R.; Ossendorp, F.; Hall, T.V.; Melief, C.J.M. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219. [Google Scholar] [CrossRef]
- Van, D.P.L.; Stork, M. Outer membrane vesicles as platform vaccine technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Perrett, K.P.; McVernon, J.; Richmond, P.C.; Marshall, H.; Nissen, M.; August, A.; Percell, S.; Toneatto, D.; Nolan, T. Immune responses to a recombinant, four-component, meningococcal serogroup B vaccine (4CMenB) in adolescents: A phase III, randomized, multicentre, lot-to-lot consistency study. Vaccine 2015, 33, 5217–5224. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, R.; Fernández, S.; Zayas, C.; Acosta, A.; Sarmiento, M.E.; Ferro, V.A.; Rosenqvist, E.; Campa, C.; Cardoso, D.; Garcia, L. Bacterial Outer Membrane Vesicles and Vaccine Applications. Front. Immunol. 2014, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Reyes, F.; Tirado, Y.; Puig, A.; Borrero, R.; Reyes, G. Immunogenicity and cross-reactivity against Mycobacterium tuberculosis;of proteoliposomes derived from Mycobacterium bovis BCG. BMC Immunol. 2013, 14, S7. [Google Scholar] [PubMed]
- Rodriguez, L.; Tirado, Y.; Reyes, F.; Puig, A.; Kadir, R.; Borrero, R.; Fernandez, S.; Reyes, G.; Alvarez, N.; Garcia, M.A. Proteoliposomes from Mycobacterium smegmatis induce immune cross-reactivity against Mycobacterium tuberculosis antigens in mice. Vaccine 2011, 29, 6236–6241. [Google Scholar] [CrossRef] [PubMed]
- Johan, H.; Philipp, O.; Richard, A.; Tatley, M.V.; Naess, L.; Aaberge, I.S.; Yvonne, G.; Anne, M.N.; Jane, O.H.; Einar, R. Vaccines against meningococcal serogroup B disease containing outer membrane vesicles (OMV): Lessons from past programs and implications for the future. Hum. Vaccines Immunother. 2013, 9, 1241–1253. [Google Scholar]
- Fernandez, S.; Fajardo, E.M.; Mandiarote, A.; Padron, M.A. A proteoliposome formulation derived from Bordetella pertussis induces;protection in two murine challenge models. BMC Immunol. 2013, 14, 1–4. [Google Scholar] [CrossRef]
- O’Donoghue, E.J.; Krachler, A.M. Mechanisms of outer membrane vesicle entry into host cells. Cell. Microbiol. 2016, 18, 1508–1517. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Fonseca, S.; Miquel-Clopes, A.; Cross, K.; Kok, K.S.; Wegmann, U.; Gil-Cordoso, K.; Bentley, E.G.; Al Katy, S.H.M.; Coombes, J.L.; et al. Bioengineering commensal bacteria-derived outer membrane vesicles for delivery of biologics to the gastrointestinal and respiratory tract. J. Extracell. Vesicles 2019, 8, 1632100. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Q.; Yi, J.; Liang, K.; Hu, B.; Zhang, X.; Curtiss, R., 3rd; Kong, Q. Outer membrane vesicles from flagellin-deficient Salmonella enterica serovar Typhimurium induce cross-reactive immunity and provide cross-protection against heterologous Salmonella challenge. Sci. Rep. 2016, 6, 34776. [Google Scholar] [CrossRef]
- Hays, M.P.; Houben, D.; Yang, Y.; Luirink, J.; Hardwidge, P.R. Immunization with Skp Delivered on Outer Membrane Vesicles Protects Mice Against Enterotoxigenic Escherichia coli Challenge. Front. Cell. Infect. Microbiol. 2018, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Marion, C.R.; Lee, J.; Sharma, L.; Park, K.S.; Lee, C.; Liu, W.; Liu, P.; Feng, J.; Gho, Y.S.; Dela Cruz, C.S. Toll-like Receptors 2 and 4 Modulate Pulmonary Inflammation and Host Factors Mediated by Outer Membrane Vesicles derived from Acinetobacter baumannii. Infect. Immun. 2019. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yao, Y.; Long, Q.; Yang, X.; Sun, W.; Liu, C.; Jin, X.; Li, Y.; Chu, X.; Chen, B.; et al. Immunization against multidrug-resistant Acinetobacter baumannii effectively protects mice in both pneumonia and sepsis models. PLoS ONE 2014, 9, e100727. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.S.; Champion, C.I.; Wu, X.Y.; Skare, J.T.; Blanco, D.R.; Miller, J.N.; Lovett, M.A. Comparison of protection in rabbits against host-adapted and cultivated Borrelia burgdorferi following infection-derived immunity or immunization with outer membrane vesicles or outer surface protein A. Infect. Immun. 2000, 68, 4189–4199. [Google Scholar] [CrossRef]
- Nieves, W.; Asakrah, S.; Qazi, O.; Brown, K.A.; Kurtz, J.; Aucoin, D.P.; McLachlan, J.B.; Roy, C.J.; Morici, L.A. A naturally derived outer-membrane vesicle vaccine protects against lethal pulmonary Burkholderia pseudomallei infection. Vaccine 2011, 29, 8381–8389. [Google Scholar] [CrossRef]
- Zurita, M.E.; Wilk, M.M.; Carriquiriborde, F.; Bartel, E.; Moreno, G.; Misiak, A.; Mills, K.H.G.; Hozbor, D. A Pertussis Outer Membrane Vesicle-Based Vaccine Induces Lung-Resident Memory CD4 T Cells and Protection Against Bordetella pertussis, Including Pertactin Deficient Strains. Front. Cell. Infect. Microbiol. 2019, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Hong, B.S.; Park, K.S.; Yoon, Y.J.; Choi, S.J.; Lee, W.H.; Roh, T.Y.; Lotvall, J.; Kim, Y.K.; Gho, Y.S. Immunization with Escherichia coli outer membrane vesicles protects bacteria-induced lethality via Th1 and Th17 cell responses. J. Immunol. 2013, 190, 4092–4102. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhao, Q.; Wen, X.; Wu, R.; Wen, Y.; Huang, X.; Huang, Y.; Yan, Q.; Han, X.; Ma, X.; et al. A trivalent Apx-fusion protein delivered by E. coli outer membrane vesicles induce protection against Actinobacillus pleuropneumoniae of serotype 1 and 7 challenge in a murine model. PLoS ONE 2018, 13, e0191286. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.; Oliaro, J.; Domigan, N.; Potter, H.; Aitken, G.; Allardyce, R.; Roake, J. Immune response to an 18-kilodalton outer membrane antigen identifies lipoprotein 20 as a Helicobacter pylori vaccine candidate. Infect. Immunol. 2000, 68, 3337–3343. [Google Scholar] [CrossRef]
- Lee, W.H.; Choi, H.I.; Hong, S.W.; Kim, K.S.; Gho, Y.S.; Jeon, S.G. Vaccination with Klebsiella pneumoniae-derived extracellular vesicles protects against bacteria-induced lethality via both humoral and cellular immunity. Exp. Mol. Med. 2015, 47, e183. [Google Scholar] [CrossRef]
- Bai, X.; Findlow, J.; Borrow, R. Recombinant protein meningococcal serogroup B vaccine combined with outer membrane vesicles. Expert Opin. Biol. Ther. 2011, 11, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Etchart, N.; Baaten, B.; Andersen, S.R.; Hyland, L.; Wong, S.Y.; Hou, S. Intranasal immunisation with inactivated RSV and bacterial adjuvants induces mucosal protection and abrogates eosinophilia upon challenge. Eur. J. Immunol. 2006, 36, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Hasegawa, H.; Ochiai, K.; Takashiba, S.; Ainai, A.; Ohnishi, M.; Watanabe, H.; Senpuku, H. Outer membrane vesicles of Porphyromonas gingivalis elicit a mucosal immune response. PLoS ONE 2011, 6, e26163. [Google Scholar] [CrossRef]
- Liu, Q.; Yi, J.; Liang, K.; Zhang, X.; Liu, Q. Outer Membrane Vesicles Derived from Salmonella Enteritidis Protect against the Virulent Wild-Type Strain Infection in a Mouse Model. J. Microbiol. Biotechnol. 2017, 27, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Alaniz, R.C.; Deatherage, B.L.; Lara, J.C.; Cookson, B.T. Membrane vesicles are immunogenic facsimiles of Salmonella typhimurium that potently activate dendritic cells, prime B and T cell responses, and stimulate protective immunity in vivo. J. Immunol. 2007, 179, 7692–7701. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.I.; Irache, J.M.; de Souza, J.; Sanchez-Gomez, S.; Gamazo, C. Nanoparticle-based vaccine for mucosal protection against Shigella flexneri in mice. Vaccine 2013, 31, 3288–3294. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Sinha, R.; Mitobe, J.; Koley, H. Development of a cost-effective vaccine candidate with outer membrane vesicles of a tolA-disrupted Shigella boydii strain. Vaccine 2016, 34, 1839–1846. [Google Scholar] [CrossRef]
- Bishop, A.L.; Tarique, A.A.; Patimalla, B.; Calderwood, S.B.; Qadri, F.; Camilli, A. Immunization of mice with vibrio cholerae outer-membrane vesicles protects against hyperinfectious challenge and blocks transmission. J. Infect. Dis. 2012, 205, 412–421. [Google Scholar] [CrossRef]
- Bartolini, E.; Ianni, E.; Frigimelica, E.; Petracca, R.; Galli, G.; Scorza, F.B.; Norais, N.; Laera, D.; Giusti, F.; Pierleoni, A. Recombinant outer membrane vesicles carrying Chlamydia muridarum HtrA induce antibodies that neutralize chlamydial infection in vitro. J. Extracell. Vesicles 2013, 2, 20181. [Google Scholar] [CrossRef]
- Muralinath, M.; Kuehn, M.J.; Roland, K.L.; Curtiss, R., 3rd. Immunization with Salmonella enterica serovar Typhimurium-derived outer membrane vesicles delivering the pneumococcal protein PspA confers protection against challenge with Streptococcus pneumoniae. Infect. Immun. 2011, 79, 887–894. [Google Scholar] [CrossRef]
- Hickey, C.A.; Kuhn, K.A.; Donermeyer, D.L.; Porter, N.T.; Jin, C.; Cameron, E.A.; Jung, H.; Kaiko, G.E.; Wegorzewska, M.; Malvin, N.P. Colitogenic Bacteroides thetaiotaomicron Antigens Access Host Immune Cells in a Sulfatase-Dependent Manner via Outer Membrane Vesicles. Cell Host Microbe 2015, 17, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Ritter, T.E.; Fajardo, O.; Matsue, H.; Anderson, R.G.; Lacey, S.W. Folate receptors targeted to clathrin-coated pits cannot regulate vitamin uptake. Proc. Natl. Acad. Sci. USA 1995, 92, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Dries, V.; Vandenbroucke, R.E.; Jones, A.T.; Joanna, R.; Joseph, D.; De Smedt, S.C.; Sanders, N.N.; Kevin, B. The use of inhibitors to study endocytic pathways of gene carriers: Optimization and pitfalls. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 561. [Google Scholar]
- Rewatkar, P.V.; Parton, R.G.; Parekh, H.S.; Parat, M.O. Are caveolae a cellular entry route for non-viral therapeutic delivery systems? Adv. Drug. Deliv. Rev. 2015, 91, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect. Immun. 2010, 78, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.W.; Kuehn, M.J.; Mason, K.M. Elicitation of epithelial cell-derived immune effectors by outer membrane vesicles of nontypeable Haemophilus influenzae. Infect. Immun. 2011, 79, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.T. Outer Membrane Vesicles (OMVs) of Gram-negative Bacteria: A Perspective Update. Front. Microbiol. 2017, 8, 1053. [Google Scholar] [CrossRef]
- Weiner, A.; Mellouk, N.; Lopez-Montero, N.; Chang, Y.Y.; Souque, C.; Schmitt, C.; Enninga, J. Macropinosomes are Key Players in Early Shigella Invasion and Vacuolar Escape in Epithelial Cells. PLoS Pathog. 2016, 12, e1005602. [Google Scholar] [CrossRef]
- Jet Phey, L.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836. [Google Scholar]
- Soderblom, T.; Oxhamre, C.; Wai, S.N.; Uhlen, P.; Aperia, A.; Uhlin, B.E.; Richter-Dahlfors, A. Effects of the Escherichia coli toxin cytolysin A on mucosal immunostimulation via epithelial Ca2+ signalling and Toll-like receptor 4. Cell. Microbiol. 2005, 7, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tan, K.; Yuan, J.; Song, K.; Li, R.; Huang, X.; Liu, Q. Flagellin-deficient outer membrane vesicles as adjuvant induce cross-protection of Salmonella Typhimurium outer membrane proteins against infection by heterologous Salmonella serotypes. Int. J. Med Microbiol. 2018, 308, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Li, R.; Huang, X.; Liu, Q. Outer Membrane Vesicles: Current Status and Future Direction of These Novel Vaccine Adjuvants. Front. Microbiol. 2018, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell. Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Chaudhuri, K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J. Biol. Chem. 2013, 288, 4299–4309. [Google Scholar] [CrossRef] [PubMed]
- Aghasadeghi, M.R.; Salmani, A.S.; Sadat, S.M.; Javadi, F.; Memarnejadian, A.; Vahabpour, R.; Zabihollahi, R.; Moshiri, A.; Siadat, S.D. Application of outer membrane vesicle of Neisseria meningitidis serogroup B as a new adjuvant to induce strongly Th1-oriented responses against HIV-1. Curr. HIV Res. 2011, 9, 630–635. [Google Scholar] [CrossRef]
- Grandi, A.; Tomasi, M.; Zanella, I.; Ganfini, L.; Caproni, E.; Fantappiè, L.; Irene, C.; Frattini, L.; Isaac, S.J.; König, E. Synergistic Protective Activity of Tumor-Specific Epitopes Engineered in Bacterial Outer Membrane Vesicles. Front. Oncol. 2017, 7, 253. [Google Scholar] [CrossRef]
- Bonnington, K.E.; Kuehn, M.J. Protein selection and export via outer membrane vesicles. Biochim. Et Biophys. Acta 2014, 1843, 1612–1619. [Google Scholar] [CrossRef]
- Kolling, G.L.; Matthews, K.R. Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl. Environ.Microbiol. 1999, 65, 1843–1848. [Google Scholar]
- Chen, Q.; Rozovsky, S.; Chen, W. Engineering multi-functional bacterial outer membrane vesicles as modular nanodevices for biosensing and bioimaging. Chem. Commun. 2017, 53, 7569–7572. [Google Scholar] [CrossRef]
- Berthiaume, F.; Leblond, M.F.; Harel, J.; Mourez, M. Growth-phase-dependent expression of the operon coding for the glycosylated autotransporter adhesin AIDA-I of pathogenic Escherichia coli. FEMS Microbiol. Lett. 2010, 311, 176–184. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.S.; Lee, S.R.; Kim, E.; Kim, M.S.; Lee, E.Y.; Gho, Y.S.; Kim, J.W.; Bishop, R.E.; Chang, K.T. Structural modifications of outer membrane vesicles to refine them as vaccine delivery vehicles. Biochim. Et Biophys. Acta 2009, 1788, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Salverda, M.L.; Meinderts, S.M.; Hamstra, H.J.; Wagemakers, A.; Hovius, J.W.; van der Ark, A.; Stork, M.; van der Ley, P. Surface display of a borrelial lipoprotein on meningococcal outer membrane vesicles. Vaccine 2016, 34, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.W.; Lindmark, B.; Söderblom, T.; Takade, A.; Westermark, M.; Oscarsson, J.; Jass, J.; Richter-Dahlfors, A.; Mizunoe, Y.; Uhlin, B.E. Vesicle-Mediated Export and Assembly of Pore-Forming Oligomers of the Enterobacterial ClyA Cytotoxin. Cell 2003, 115, 25–35. [Google Scholar]
- Kim, J.Y.; Doody, A.M.; Chen, D.J.; Cremona, G.H.; Shuler, M.L.; Putnam, D.; Delisa, M.P. Engineered Bacterial Outer Membrane Vesicles with Enhanced Functionality. J. Mol. Biol. 2008, 380, 51–66. [Google Scholar] [CrossRef]
- Chen, D.J.; Nikolaus, O.; Metzger, S.M.; Elizabeth, B.; Doody, A.M.; Delisa, M.P.; David, P. Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc. Natl. Acad. Sci. USA 2010, 107, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, K.; Daleke-Schermerhorn, M.H.; Jong, W.S.; ten Hagen-Jongman, C.M.; van Opzeeland, F.; Simonetti, E.; Luirink, J.; de Jonge, M.I. Salmonella outer membrane vesicles displaying high densities of pneumococcal antigen at the surface offer protection against colonization. Vaccine 2015, 33, 2022–2029. [Google Scholar] [CrossRef]
- Daleke-Schermerhorn, M.H.; Felix, T.; Soprova, Z.; Ten Hagen-Jongman, C.M.; Vikstrom, D.; Majlessi, L.; Beskers, J.; Follmann, F.; de Punder, K.; van der Wel, N.N.; et al. Decoration of outer membrane vesicles with multiple antigens by using an autotransporter approach. Appl. Environ. Microbiol. 2014, 80, 5854–5865. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.S.; Daleke-Schermerhorn, M.H.; Vikstrom, D.; Ten Hagen-Jongman, C.M.; de Punder, K.; van der Wel, N.N.; van de Sandt, C.E.; Rimmelzwaan, G.F.; Follmann, F.; Agger, E.M.; et al. An autotransporter display platform for the development of multivalent recombinant bacterial vector vaccines. Microb. Cell Factories 2014, 13, 162. [Google Scholar] [CrossRef]
- Wang, S.; Huang, W.; Li, K.; Yao, Y.; Yang, X.; Bai, H.; Sun, W.; Liu, C.; Ma, Y. Engineered outer membrane vesicle is potent to elicit HPV16E7-specific cellular immunity in a mouse model of TC-1 graft tumor. Int. J Nanomed. 2017, 12, 6813–6825. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release Soc. 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Tahmasbi Rad, A.; Chen, C.W.; Aresh, W.; Xia, Y.; Lai, P.S.; Nieh, M.P. Combinational Effects of Active Targeting, Shape, and Enhanced Permeability and Retention for Cancer Theranostic Nanocarriers. ACS Appl. Mater. Interfaces 2019, 11, 10505–10519. [Google Scholar] [CrossRef] [PubMed]
- Vipul, G.; Sunghyun, K.; Sang-Hyun, K.; Jung Joon, M.; Choy, H.E.; Chang, K.S.; Sangyong, J. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano 2014, 8, 1525–1537. [Google Scholar]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res. 1991, 105, 3–11. [Google Scholar]
- Wallecha, A.; Malinina, I.; Ramos, K.; Singh, R. Abstract 5241: Development of a single Listeria monocytogene-based vaccine simultaneously targeting two antigens. Cancer Res. 2012, 72, 5241. [Google Scholar]
- Wang, S.; Kong, Q.; Iii, R.C. New technologies in developing recombinant attenuated Salmonella vaccine vectors. Microb. Pathog. 2013, 58, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, T.; Xavier, C.; Yan, W.; Benoit, P.; Audrey, L.G.L. Live-attenuated bacteria as a cancer vaccine vector. Expert Rev. Vaccines 2013, 12, 1139–1154. [Google Scholar]
- Nicholas, A.; Jeon, H.; Selasi, G.N.; Na, S.H.; Kwon, H.I.; Kim, Y.J.; Choi, C.W.; Kim, S.I.; Lee, J.C. Clostridium difficile-derived membrane vesicles induce the expression of pro-inflammatory cytokine genes and cytotoxicity in colonic epithelial cells in vitro. Microb. Pathog. 2017, 107, 6–11. [Google Scholar] [CrossRef]
- Kim, J.H.; Jeun, E.J.; Hong, C.P.; Kim, S.H.; Jang, M.S.; Lee, E.J.; Moon, S.J.; Yun, C.H.; Im, S.H.; Jeong, S.G.; et al. Extracellular vesicle-derived protein from Bifidobacterium longum alleviates food allergy through mast cell suppression. J. Allergy Clin. Immunol. 2016, 137, 507–516.e508. [Google Scholar] [CrossRef]
- Kim, O.Y.; Park, H.T.; Nth, D.; Choi, S.J.; Lee, J.; Kim, J.H.; Lee, S.W.; Gho, Y.S. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat. Commun. 2017, 8, 626. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Le’Negrate, G.; Krajewska, M.; Reed, J.C. IL-18-producing Salmonella inhibit tumor growth. Cancer Gene Ther. 2008, 15, 787. [Google Scholar] [CrossRef] [PubMed]
- Markus, L.; Gaelle, L.N.; Maryla, K.; Reed, J.C. Attenuated Salmonella engineered to produce human cytokine LIGHT inhibit tumor growth. Proc. Natl. Acad. Sci. USA 2007, 104, 12879–12883. [Google Scholar]
- Loeffler, M.; Le’Negrate, G.; Krajewska, M.; Reed, J.C. Salmonella typhimurium engineered to produce CCL21 inhibit tumor growth. Cancer Immunol. Immunother. 2009, 58, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.; Arenas, R.B.; Forbes, N.S. Tumour-targeted delivery of TRAIL using Salmonella typhimurium enhances breast cancer survival in mice. Br. J. Cancer 2009, 101, 1683. [Google Scholar] [CrossRef]
- Loeffler, M.; Le’Negrate, G.M.; Reed, J. Inhibition of tumor growth using salmonella expressing Fas ligand. J. Natl. Cancer. Inst. 2008, 100, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Bereta, M.; Hayhurst, A.; Gajda, M.; Chorobik, P.; Targosz, M.; Marcinkiewicz, J.; Kaufman, H.L. Improving tumor targeting and therapeutic potential of Salmonella VNP20009 by displaying cell surface CEA-specific antibodies. Vaccine 2007, 25, 4183–4192. [Google Scholar] [CrossRef]
- Quintero, D.; Carrafa, J.; Vincent, L.; Bermudes, D. EGFR-targeted chimeras of Pseudomonas ToxA released into the extracellular milieu by attenuated Salmonella selectively kill tumor cells. Biotechnol. Bioeng. 2016, 113, 2698–2711. [Google Scholar] [CrossRef]
- King, I.; Bermudes, D.; Lin, S.; Belcourt, M.; Pike, J.; Troy, K.; Le, T.; Ittensohn, M.; Mao, J.; Lang, W. Tumor-targeted salmonella expressing Cytosine deaminase as an anticancer agent. Hum. Gene Ther. 2002, 13, 1225–1233. [Google Scholar] [CrossRef]
- Zhao, C.; He, J.; Cheng, H.; Zhu, Z.; Xu, H. Enhanced therapeutic effect of an antiangiogenesis peptide on lung cancer in vivo combined with salmonella VNP20009 carrying a Sox2 shRNA construct. J. Exp. Clin. Cancer Res. 2016, 35, 107. [Google Scholar] [CrossRef]
- Blache, C.A.; Manuel, E.R.; Kaltcheva, T.I.; Wong, A.N.; Ellenhorn, J.D.I.; Blazar, B.R.; Diamond, D.J. Systemic delivery of Salmonella typhimurium transformed with IDO shRNA enhances intratumoral vector colonization and suppresses tumor growth. Cancer Res. 2011, 72, 6447–6456. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, B.S.; Banton, K.L.; Frykman, N.L.; Leonard, A.S.; Saltzman, D.A. Attenuated Salmonella typhimurium with IL-2 Gene Reduces Pulmonary Metastases in Murine Osteosarcoma. Clin. Orthop. Relat. Res. 2008, 466, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.; Park, Y.C.; Kim, J.; Yang, S.C.; Byeon, J.H.; Min, S.H.; Park, S.; Yoo, Y.; Yong, K.P.; Kim, B.M. Application of genetically engineered Salmonella typhimurium for interferon-gamma–induced therapy against melanoma. Eur. J. Cancer 2017, 70, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Hyung-Seok, K.; Jung-Min, H.; Yeongjin, H.; Choy, H.E.; Jung-Joon, M. Genetically engineered Salmonella typhimurium as an imageable therapeutic probe for cancer. Cancer Res. 2010, 70, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Jae-Ho, J.; Kwangsoo, K.; Daejin, L.; Kwangjoon, J.; Yeongjin, H.; Nguyen, V.H.; Tae-Hyoung, K.; Sangryeol, R.; Jeong, A.L.; Jae, K., II. Anti-tumoral effect of the mitochondrial target domain of Noxa delivered by an engineered Salmonella typhimurium. PLoS ONE 2014, 9, e80050. [Google Scholar]
- Zheng, J.H.; Nguyen, V.H.; Jiang, S.N.; Park, S.H.; Tan, W.; Hong, S.H.; Shin, M.G.; Chung, I.J.; Hong, Y.; Bom, H.S. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med. 2017, 9, eaak9537. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.E.; Aida, P.; Ana, M.; Ario, D.M.; Maria, R. Salmonella engineered to express CD20-targeting antibodies and a drug-converting enzyme can eradicate human lymphomas. Blood 2013, 122, 705–714. [Google Scholar] [CrossRef]
- Yoon, W.; Ji, H.C.; Kim, S.; Yong, K.P. Engineered Salmonella typhimurium expressing E7 fusion protein, derived from human papillomavirus, inhibits tumor growth in cervical tumor-bearing mice. Biotechnol. Lett. 2014, 36, 349–356. [Google Scholar] [CrossRef]
- Yuhua, L.; Kunyuan, G.; Hui, C.; Yongmei, X.; Chaoyang, S.; Xun, T.; Daming, R. Oral cytokine gene therapy against murine tumor using attenuated Salmonella typhimurium. Int. J. Cancer J. Int. Du Cancer 2010, 94, 438–443. [Google Scholar] [CrossRef]
- Caroline, A.; Fernanda, S.; Mark, S.; Piero, M.; Marcelo, F.; Miguel Angel, M.; Jose Alejandro, C. Live attenuated Salmonella as a vector for oral cytokine gene therapy in melanoma. J. Gene Med. 2010, 9, 416–423. [Google Scholar]
- Cao, H.D.; Yang, Y.X.; Lin, L.; Liu, S.N.; Wang, P.L.; Tao, X.H.; Wang, L.J.; Xiang, T.X. Attenuated Salmonella typhimurium carrying TRAIL and VP3 genes inhibits the growth of gastric cancer cells in vitro and in vivo. Tumori 2010, 96, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Yu, B.; Cai, C.H.; Huang, W.; Zheng, B.J.; Smith, D.K.; Huang, J.D. Combined prokaryotic–eukaryotic delivery and expression of therapeutic factors through a primed autocatalytic positive-feedback loop. J. Control. Release 2016, 222, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Jianxin, Y.; Ling, L.; Yuanting, Z.; Xueguang, Z.; Daming, R.; Weichang, C. Recombinant Salmonella-based 4-1BBL vaccine enhances T cell immunity and inhibits the development of colorectal cancer in rats: In vivo effects of vaccine containing 4-1BBL. J. Biomed. Sci. 2013, 20, 8. [Google Scholar]
- Elisa, B.; Rocio, S.; Nicole, H.; Oliver, H.; Alexander, S.; Tahir, D.; Stephan, L.; Zenclussen, A.C.; Holger, C.; Lode, H.N. Salmonella SL7207 application is the most effective DNA vaccine delivery method for successful tumor eradication in a murine model for neuroblastoma. Cancer Lett. 2013, 331, 167–173. [Google Scholar]
- Shu, G.Z.; Chen, Y.; Zhi, P.W. Orally Administered DNA Vaccine Delivery by Attenuated Salmonellatyphimurium Targeting Fetal Liver Kinase 1 Inhibits Murine Lewis LungCarcinoma Growth and Metastasis. Biol. Pharm. Bull. 2010, 33, 174–182. [Google Scholar]
- Ahmad, S.; Casey, G.; Cronin, M.; Rajendran, S.; Sweeney, P.; Tangney, M.; O’Sullivan, G.C. Induction of Effective Antitumor Response After Mucosal Bacterial Vector Mediated DNA Vaccination With Endogenous Prostate Cancer Specific Antigen. J. Urol. 2011, 186, 687–693. [Google Scholar] [CrossRef]
- Zhou, J.H.; Chen, Y.C.; Li, T. Tumor-targeting Salmonella typhimurium, a natural tool for activation of prodrug 6MePdR and their combination therapy in murine melanoma model. Appl. Microbiol. Biotechnol. 2013, 97, 4393–4401. [Google Scholar]
- Nan, Y.; Zhu, X.; Chen, L.; Li, S.; Ren, D. Oral administration of attenuated S. typhimurium carrying shRNA-expressing vectors as a cancer therapeutic. Cancer Biol. Ther. 2008, 7, 145–151. [Google Scholar]
- Kim, K.; Jeong, J.H.; Lim, D.; Hong, Y.; Lim, H.J.; Kim, G.J.; Shin, S.R.; Lee, J.J.; Yun, M.; Harris, R.A. L-Asparaginase delivered by Salmonella typhimurium suppresses solid tumors. Mol. Ther. Oncol. 2015, 2, 15007. [Google Scholar] [CrossRef]
- Park, S.H.; Zheng, J.H.; Nguyen, V.H.; Jiang, S.N.; Kim, D.Y.; Szardenings, M.; Min, J.H.; Hong, Y.; Choy, H.E.; Min, J.J. RGD Peptide Cell-Surface Display Enhances the Targeting and Therapeutic Efficacy of AttenuatedSalmonella-mediated Cancer Therapy. Theranostics 2016, 6, 1672–1682. [Google Scholar] [CrossRef]
- Shi, L.; Yu, B.; Cai, C.H.; Huang, J.D. Angiogenic inhibitors delivered by the type III secretion system of tumor-targeting Salmonella typhimurium safely shrink tumors in mice. AMB Express 2016, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Li, Y.; Zhao, T.; Li, X.; Hu, J.; Yin, D.; Guo, B.; Kopecko, D.J.; Zhao, X.; Zhang, L. Antitumor effects of Stat3-siRNA and endostatin combined therapies, delivered by attenuated Salmonella, on orthotopically implanted hepatocarcinoma. Cancer Immunol. Immunother. 2012, 61, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Guosheng, X.; Husseiny, M.I.; Liping, S.; Anat, E.E.; Shackleford, G.M.; Seeger, R.C.; Daniela, J.C.; Michael, H.; Metelitsa, L.S. Novel cancer vaccine based on genes of Salmonella pathogenicity island 2. Int. J. Cancer 2010, 126, 2622–2634. [Google Scholar]
- Al-Ramadi, B.K.; Fernandez-Cabezudo, M.J.; El-Hasasna, H.; Al-Salam, S.; Bashir, G.; Chouaib, S. Potent anti-tumor activity of systemically-administered IL2-expressing Salmonella correlates with decreased angiogenesis and enhanced tumor apoptosis. Clin. Immunol. 2009, 130, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lewēn, S.; Zhou, H.; Hu, H.; Cheng, T.; Markowitz, D.; Reisfeld, R.A.; Xiang, R.; Luo, Y. A Legumain-based minigene vaccine targets the tumor stroma and suppresses breast cancer growth and angiogenesis. Cancer Immunol. Immunother. 2008, 57, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Roider, E.; Jellbauer, S.; Köhn, B.; Berchtold, C.; Partilla, M.; Busch, D.H.; Rüssmann, H.; Panthel, K. Invasion and destruction of a murine fibrosarcoma by Salmonella -induced effector CD8 T cells as a therapeutic intervention against cancer. Cancer Immunol. Immunother. 2011, 60, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, V.; Seavey, M.M.; Maciag, P.C.; Rivera, S.; Wallecha, A. Development of a live and highly attenuated Listeria monocytogenes-based vaccine for the treatment of Her2/neu-overexpressing cancers in human. Cancer Gene Ther. 2011, 18, 53. [Google Scholar] [CrossRef]
- Seavey, M.; Maciag, P.R.N.; Sewell, D.; Paterson, Y. An anti-vascular endothelial growth factor receptor 2/fetal liver kinase-1 Listeria monocytogenes anti-angiogenesis cancer vaccine for the treatment of primary and metastatic Her-2/neu+ breast tumors in a mouse model. J. Immunol. 2009, 182, 5537–5546. [Google Scholar] [CrossRef]
- Shahabi, V.; Reyes-Reyes, M.; Wallecha, A.; Rivera, S.; Paterson, Y.; Maciag, P. Development of a Listeria monocytogenes based vaccine against prostate cancer. Cancer Immunol. Immunother. 2008, 57, 1301. [Google Scholar] [CrossRef]
- Le, D.T.; Andrea, W.G.; Vincent, P.; Greten, T.F.; Todd, C.; Gregory, S.; Michael, M.; Herbert, Z.; Deirdre, C.; Fine, R.L. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2016, 33, 1325–1333. [Google Scholar] [CrossRef]
- Crittenden, M.; Bahjat, K.S.; Li, R.; Gore, P.; Fountain, C.; Hanson, B.; Skoble, J.; Lauer, P.; Murphy, A.L.; Dubensky, T. Phase I study of safety and immunogenicity of ADU-623, a live-attenuated listeria monocytogenes vaccine (ΔactA/ΔinlB) expressing EGFRVIII and NY-ESO-1, in patients with who grade III/IV astrocytomas. J. Immunother. Cancer 2015, 3, 1–2. [Google Scholar] [CrossRef]
- Yang, J.; Zhuo, D.C.; Xudong, Z.; Chen, B. Attenuated Listeria monocytogenes as a cancer vaccine vector for the delivery of CD24, a biomarker for hepatic cancer stem cells. Cell. Mol. Immunol. 2014, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.J.; Van Zijl, P.; Fox, M.E.; Mauchline, M.L.; Giaccia, A.J.; Minton, N.P.; Brown, J.M. Anaerobic bacteria as a gene delivery system that is controlled by the tumor microenvironment. Gene Ther. 1997, 4, 791. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Landuyt, A.W.; Nuyts, S.; Van, M.L.; Lambin, P.; Anné, J. Clostridium as a tumor-specific delivery system of therapeutic proteins. Cancer Detect. Prev. 2001, 25, 548–557. [Google Scholar] [PubMed]
- Yazawa, K.; Fujimori, M.J.; Kano, Y.; Taniguchi, S. Bifidobacterium longum as a delivery system for cancer gene therapy: Selective localization and growth in hypoxic tumors. Cancer Gene Ther. 2000, 7, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Oda, T.; Saito, H.; Araki, A.; Gonoi, R.; Shigemura, K.; Hashii, Y.; Katayama, T.; Fujisawa, M.; Shirakawa, T. Development of oral cancer vaccine using recombinant Bifidobacterium displaying Wilms’ tumor 1 protein. Cancer Immunol. Immunother. 2017, 66, 787–798. [Google Scholar] [CrossRef]
- Shirakawa, T.; Kitagawa, K. Antitumor effect of oral cancer vaccine with Bifidobacterium delivering WT1 protein to gut immune system is superior to WT1 peptide vaccine. Hum. Vaccin. Immunother. 2018, 14, 159–162. [Google Scholar] [CrossRef]
- Hu, B.; Kou, L.; Li, C.; Zhu, L.P.; Fan, Y.R.; Wu, Z.W.; Wang, J.J.; Xu, G.X. Bifidobacterium longum as a delivery system of TRAIL and endostatin cooperates with chemotherapeutic drugs to inhibit hypoxic tumor growth. Cancer Gene Ther. 2009, 16, 655. [Google Scholar] [CrossRef]
- Xi, L.; Geng-Feng, F.; Yan-Rong, F.; Wen-Hua, L.; Xin-Juan, L.; Jian-Jun, W.; Gen-Xing, X. Bifidobacterium adolescentis as a delivery system of endostatin for cancer gene therapy: Selective inhibitor of angiogenesis and hypoxic tumor growth. Cancer Gene Ther. 2003, 10, 105. [Google Scholar]
- Zhu, H.; Li, Z.; Mao, S.; Ma, B.; Zhou, S.; Deng, L.; Liu, T.; Cui, D.; Zhao, Y.; He, J. Antitumor effect of sFlt-1 gene therapy system mediated by Bifidobacterium Infantis on Lewis lung cancer in mice. Cancer Gene Ther. 2011, 18, 884–896. [Google Scholar] [CrossRef]
- Derouazi, M.; Yan, W.; Marlu, R.; Epaulard, O.; Mayol, J.F.; Pasqual, N.; Gouellec, A.L.; Polack, B.; Toussaint, B. Optimal epitope composition after antigen screening using a live bacterial delivery vector. Bioeng. Bugs 2010, 1, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gouëllec, A.L.; Chaker, H.; Asrih, H.; Polack, B.; Toussaint, B. Optimization of antitumor immunotherapy mediated by type III secretion system-based live attenuated bacterial vectors. J. Immunother. 2012, 35, 223. [Google Scholar] [CrossRef] [PubMed]
- St Jean, A.T.; Swofford, C.A.; Panteli, J.T.; Brentzel, Z.J.; Forbes, N.S. Bacterial delivery of Staphylococcus aureus α-hemolysin causes regression and necrosis in murine tumors. Mol. Ther. 2014, 22, 1266–1274. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Fang, Z.; Li, R.; Huang, X.; Liu, Q. Design of Outer Membrane Vesicles as Cancer Vaccines: A New Toolkit for Cancer Therapy. Cancers 2019, 11, 1314. https://doi.org/10.3390/cancers11091314
Zhang Y, Fang Z, Li R, Huang X, Liu Q. Design of Outer Membrane Vesicles as Cancer Vaccines: A New Toolkit for Cancer Therapy. Cancers. 2019; 11(9):1314. https://doi.org/10.3390/cancers11091314
Chicago/Turabian StyleZhang, Yingxuan, Zheyan Fang, Ruizhen Li, Xiaotian Huang, and Qiong Liu. 2019. "Design of Outer Membrane Vesicles as Cancer Vaccines: A New Toolkit for Cancer Therapy" Cancers 11, no. 9: 1314. https://doi.org/10.3390/cancers11091314
APA StyleZhang, Y., Fang, Z., Li, R., Huang, X., & Liu, Q. (2019). Design of Outer Membrane Vesicles as Cancer Vaccines: A New Toolkit for Cancer Therapy. Cancers, 11(9), 1314. https://doi.org/10.3390/cancers11091314