Detection of Clinical Mesenchymal Cancer Cells from Bladder Wash Urine for Real-Time Detection and Prognosis

 ,
,  ,
,
Abstract
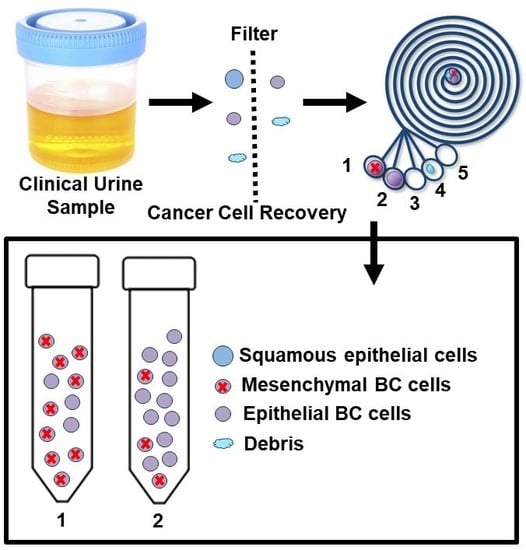
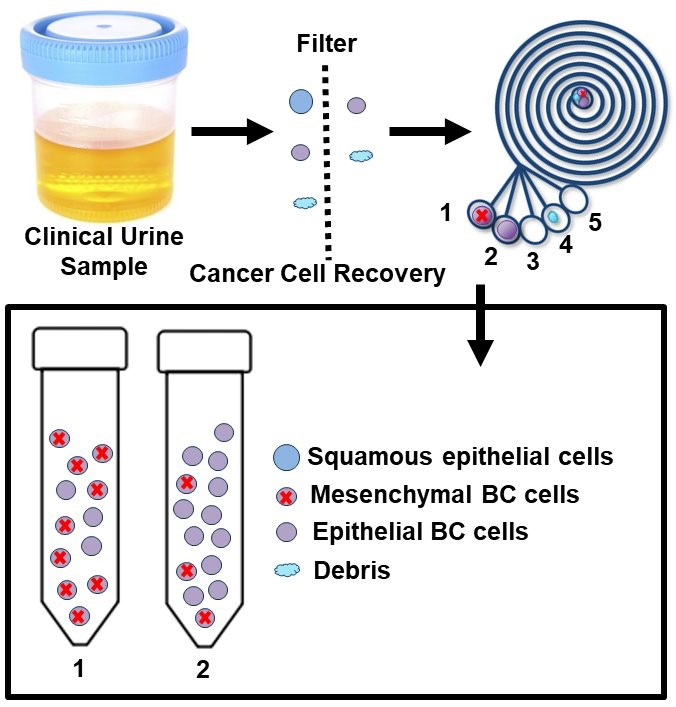
1. Introduction
2. Results
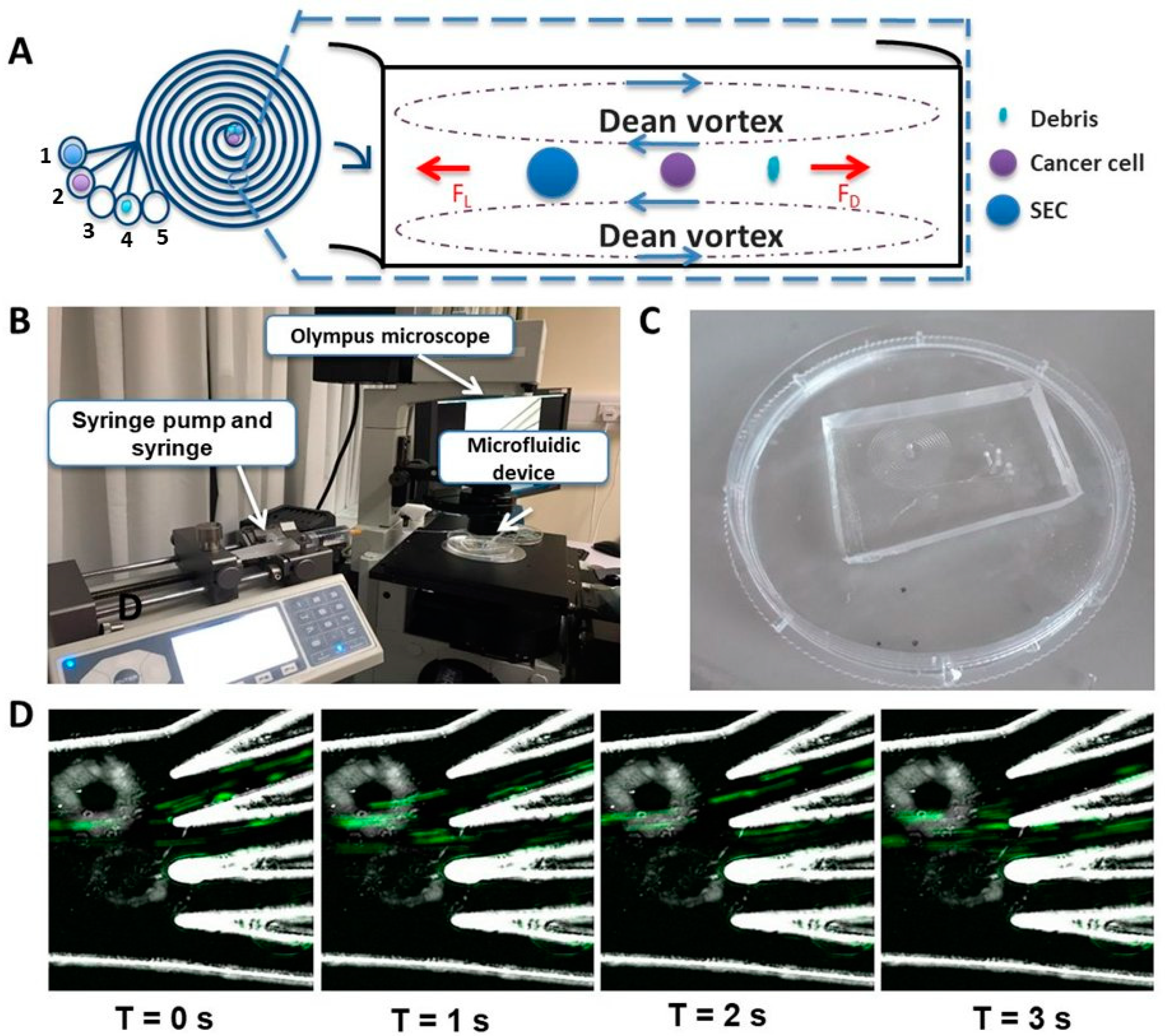
2.1. Working Principle and Design of the ES Microfluidic Enrichment Device
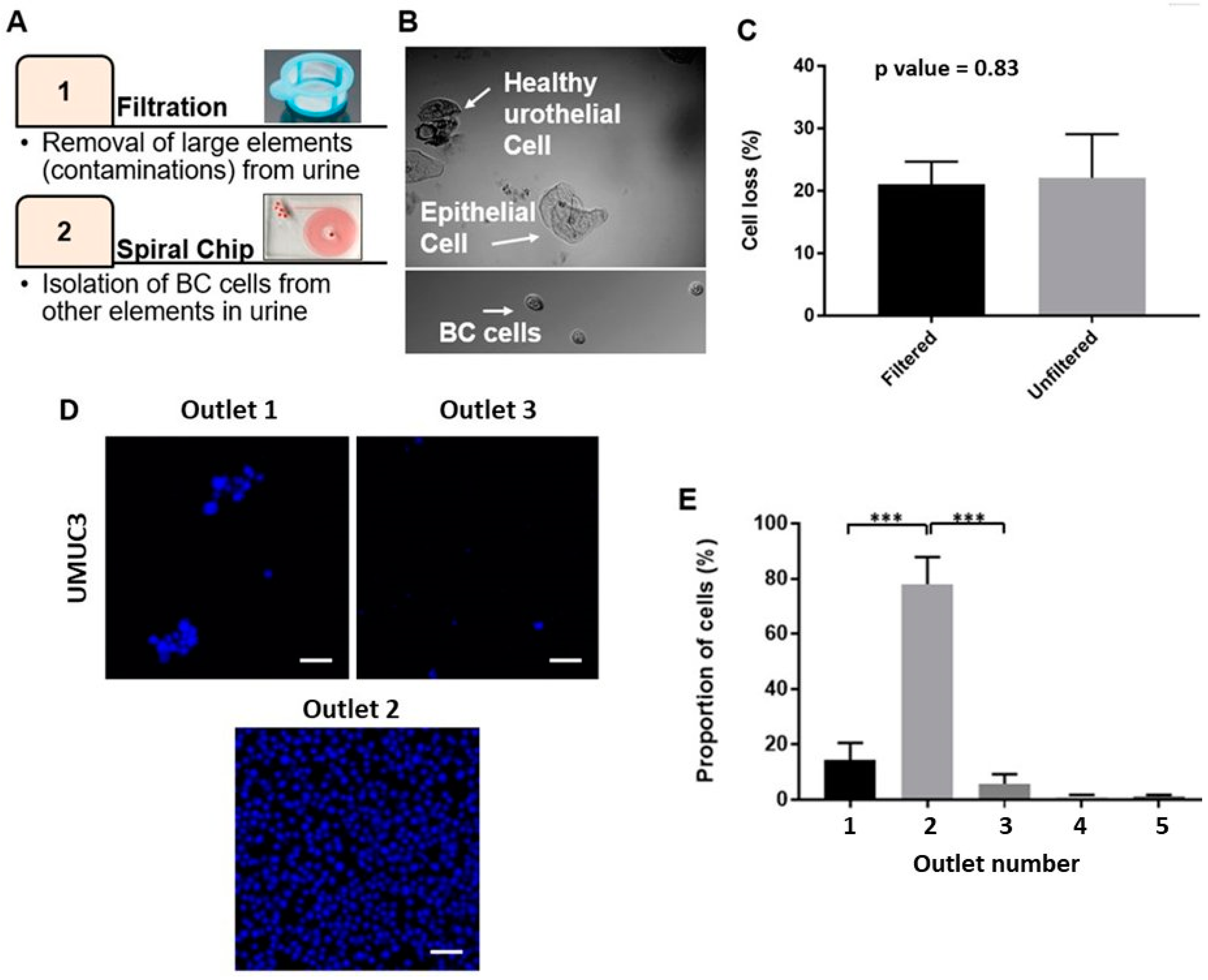
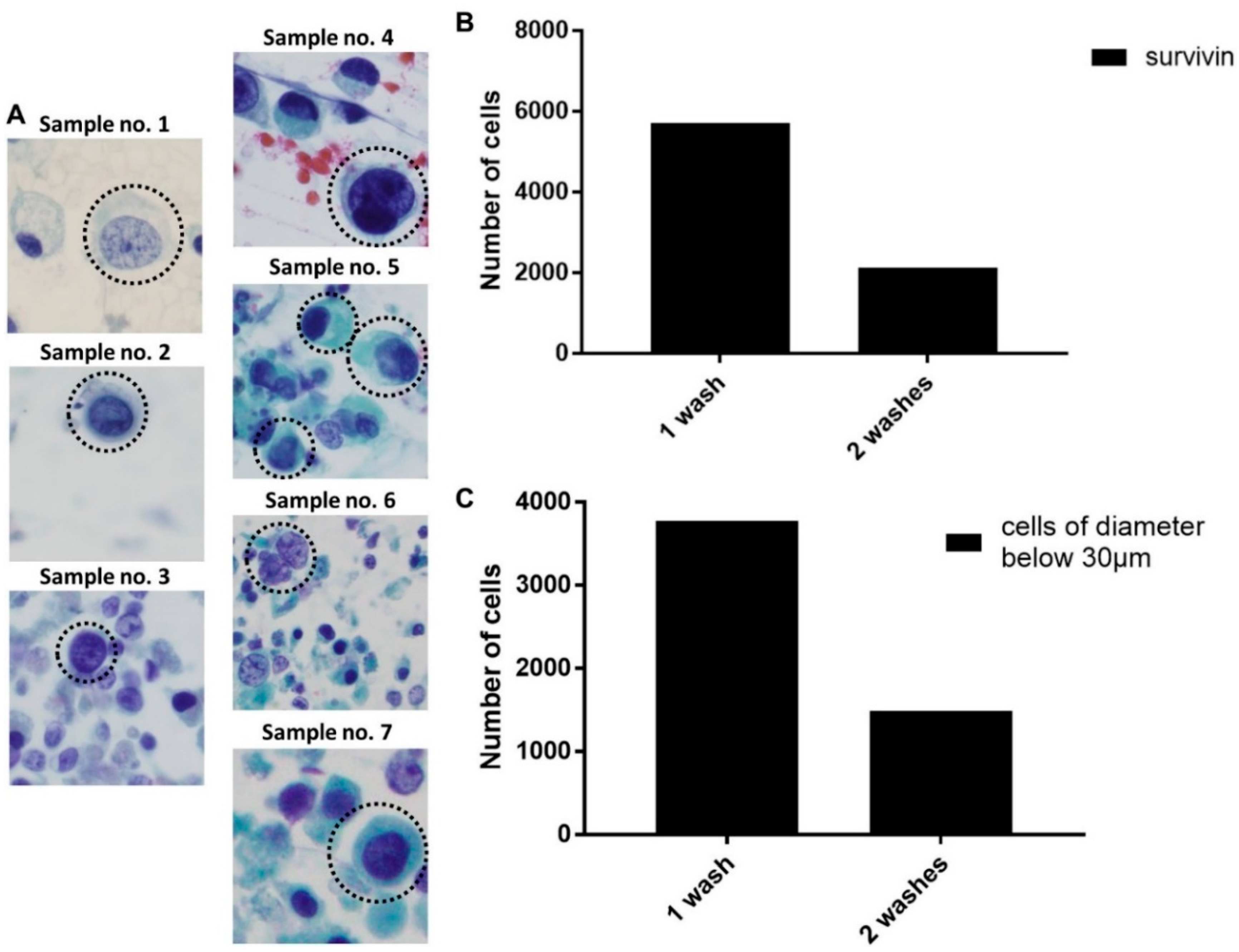
2.2. Optimizing the Efficacy of EBCCs Capture
2.3. Performance of Bladder Cancer Cells Enrichment Platform
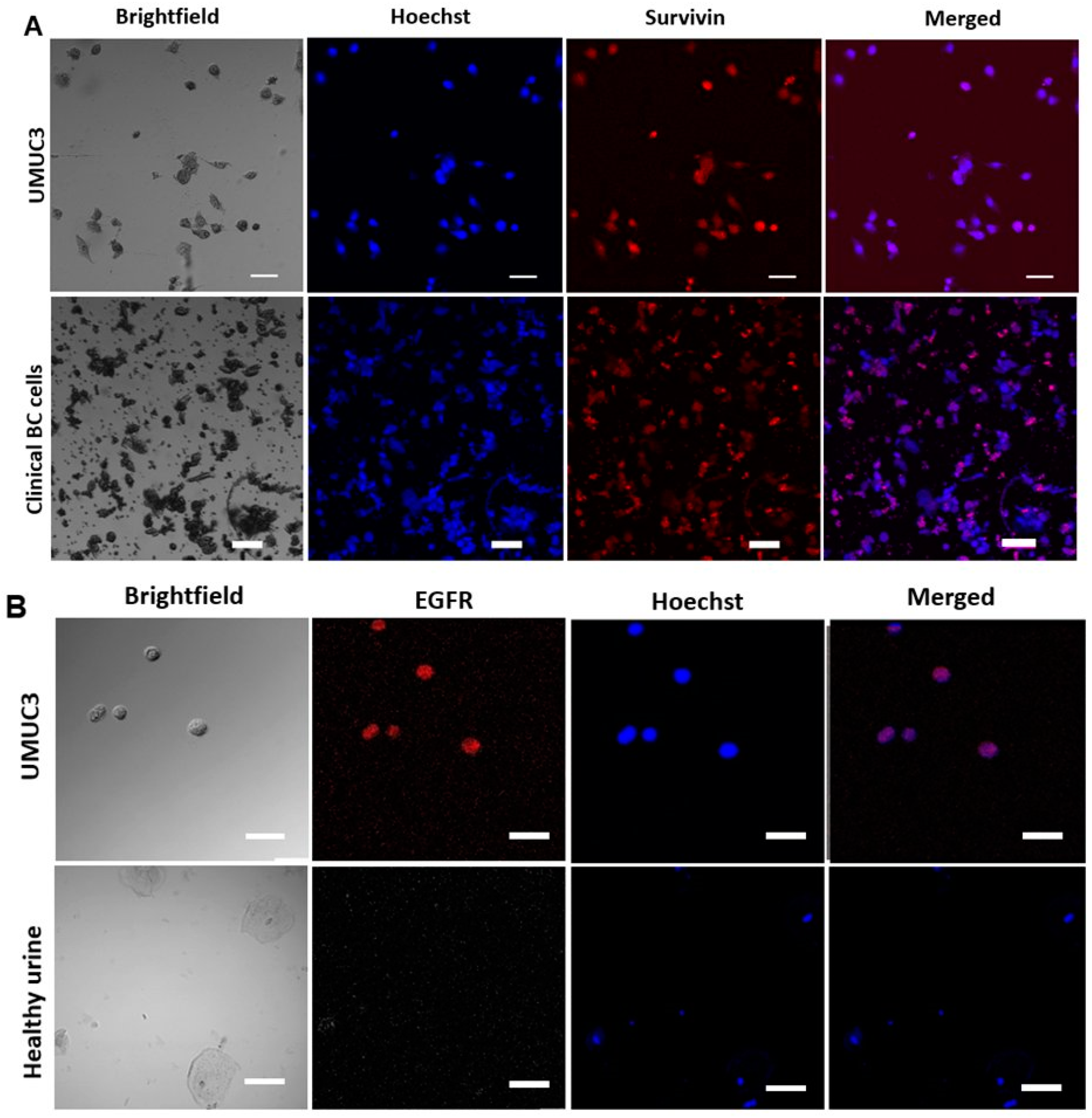
2.4. Detection of EBCCs from Spiked Cell Line Samples and Clinical Patients
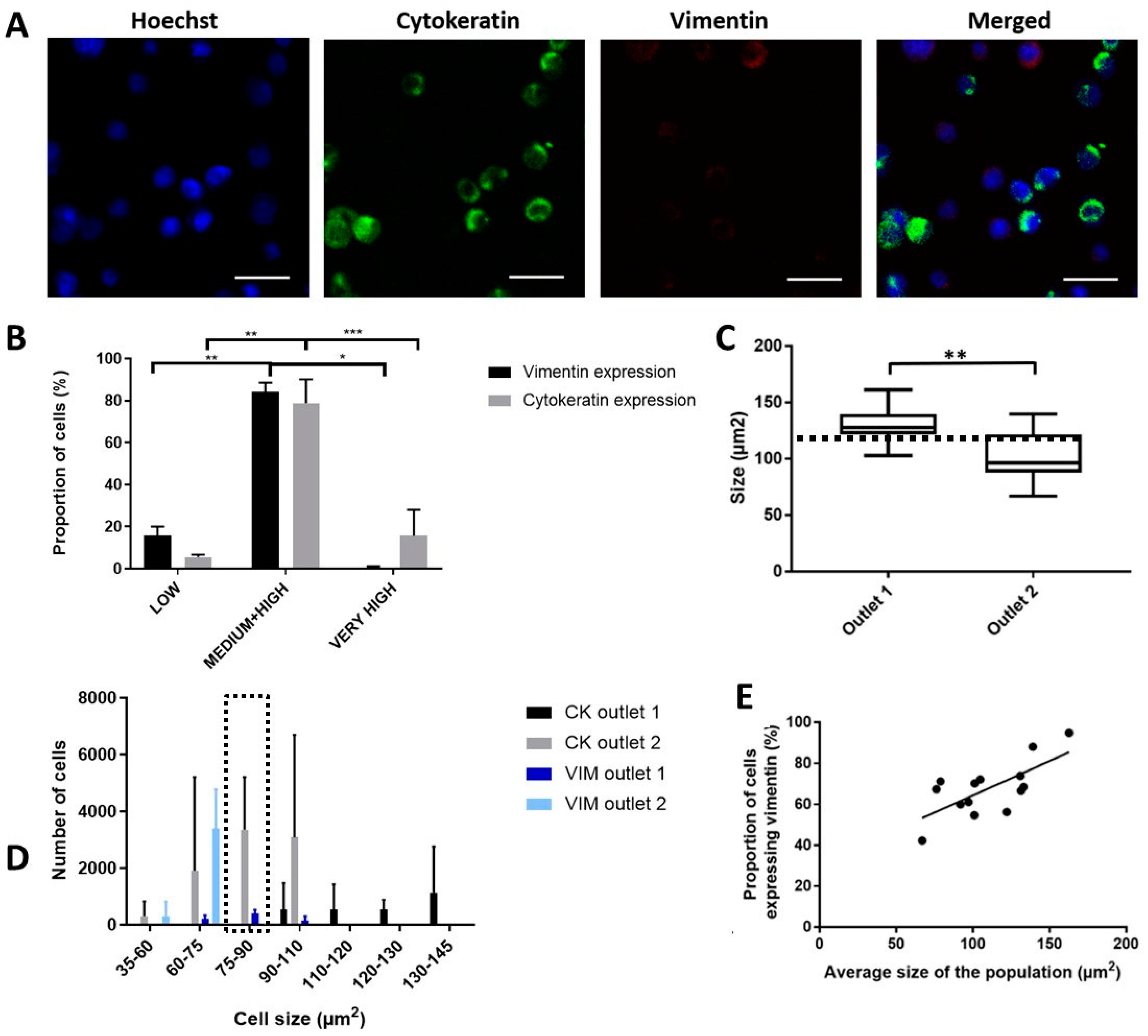
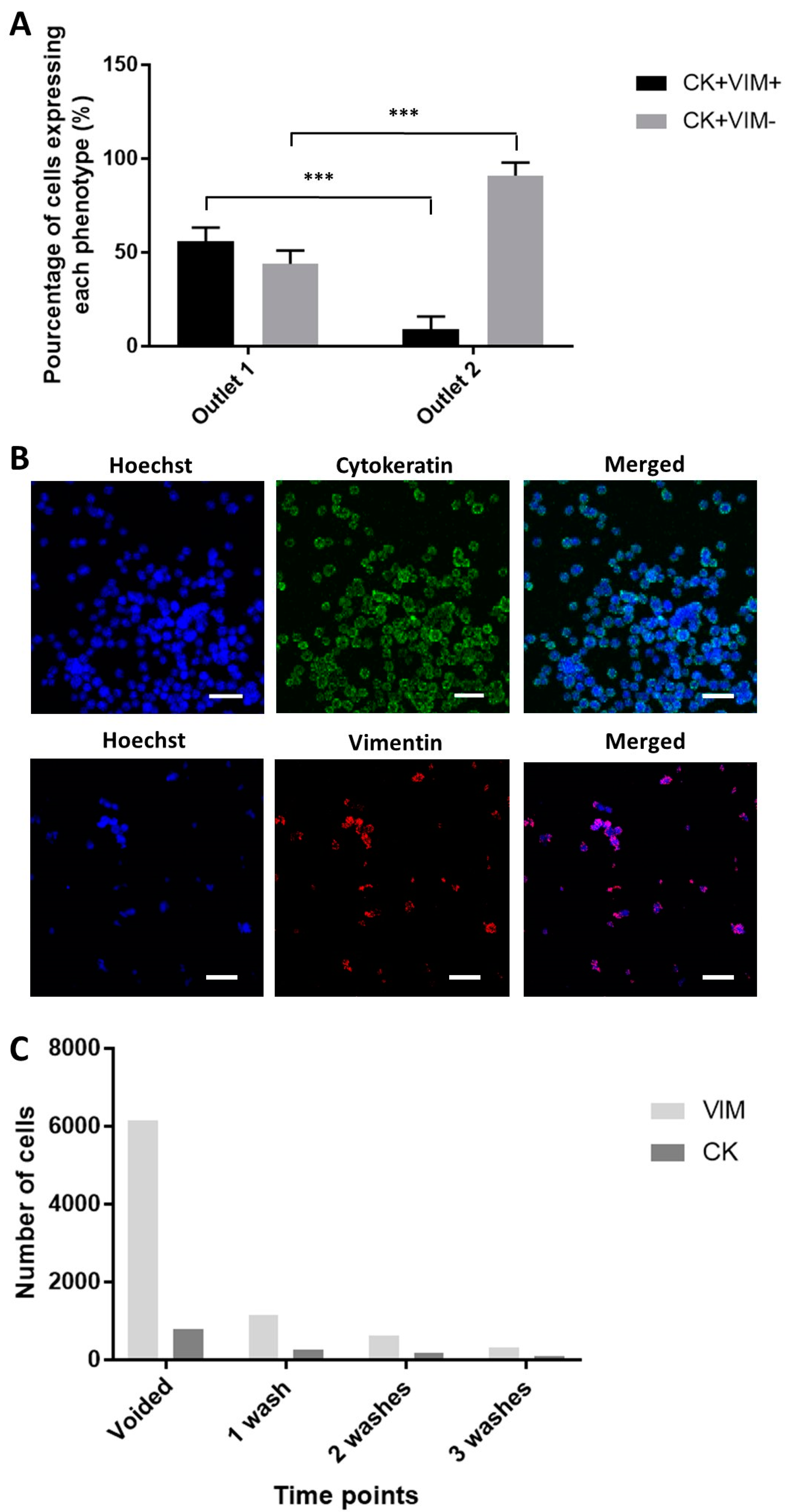
2.5. Enhanced Recovery of Clinical Mesenchymal Bladder Cancer Cells
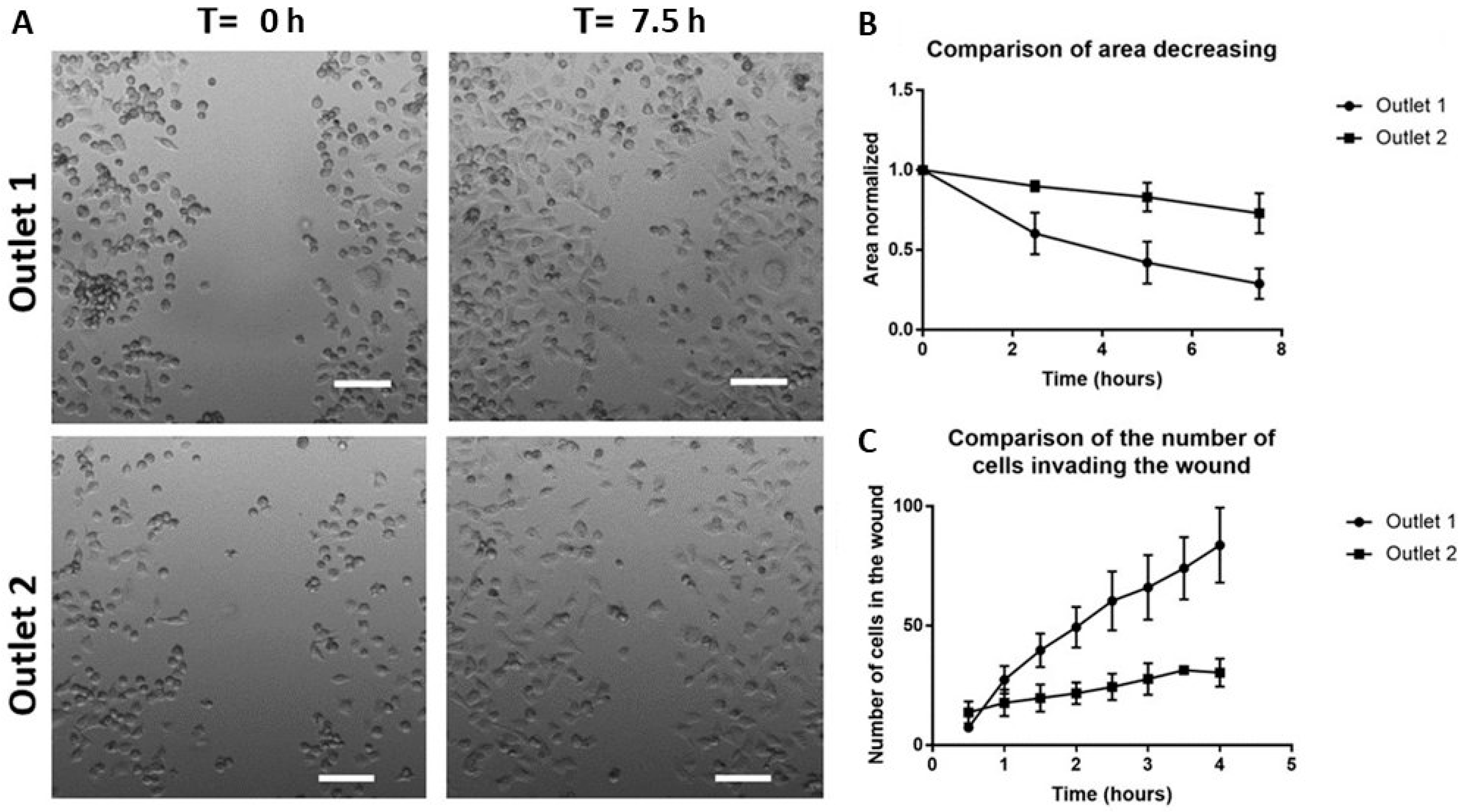
2.6. Functional Characterization of Sorted Mesenchymal Bladder Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Device Fabrication and Characterization
4.2. Cell Culture
4.3. Characterisation of Sorted UMUC3 Cells
4.4. Processing of Clinical Samples
4.5. Immunostaining of Sorted Cells
4.6. Imaging of Samples and Cell Counting
4.7. Wound Scratch Test
4.8. Statistical Summary
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Bladder Cancer |
| EBCCs | Exfoliated Bladder Cancer Cells |
| EGFR | Epidermal Growth Factor Receptor |
| SECs | Squamous Epithelial Cells |
| TURBT | Transurethral Resection of the Bladder Tumor |
| MIBC | Muscle-Invasive Bladder Cancer |
| NMBIC | Non-Muscle Invasive Bladder Cancer |
| PDMS | Polydimethylsiloxane |
| CTC | Circulating Tumor Cells |
| PBS | Phosphate Buffer Saline |
| EMT | Epithelial to Mesenchymal Transition |
| CSC | Cancer Stem Cell |
References
- Cumberbatch, M.G.K.; Jubber, I.; Black, P.C.; Esperto, F.; Figueroa, J.D.; Kamat, A.M.; Kiemeney, L.; Lotan, Y.; Pang, K.; Silverman, D.T.; et al. Epidemiology of Bladder Cancer: A Systematic Review and Contemporary Update of Risk Factors in 2018. Eur. Urol. 2018, 74, 784–795. [Google Scholar] [CrossRef]
- Andersson, E.; Steven, K.; Guldberg, P. Size-based enrichment of exfoliated tumor cells in urine increases the sensitivity for DNA-based detection of bladder cancer. PLoS ONE 2014, 9, e94023. [Google Scholar] [CrossRef]
- Falke, J.; Witjes, J.A. Contemporary management of low-risk bladder cancer. Nat. Rev. Urol. 2011, 8, 42–49. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.; Song, H.; Sohn, K.Y.; Jeon, M.; Han, K.H. Microfluidic technologies for circulating tumor cell isolation. Analyst 2018, 143, 2936–2970. [Google Scholar] [CrossRef]
- Khoo, B.L.; Grenci, G.; Lim, J.S.Y.; Lim, Y.P.; Fong, J.; Yeap, W.H.; Bin Lim, S.; Chua, S.L.; Wong, S.C.; Yap, Y.S.; et al. Low-dose anti-inflammatory combinatorial therapy reduced cancer stem cell formation in patient-derived preclinical models for tumour relapse prevention. Br. J. Cancer 2019, 120, 407–423. [Google Scholar] [CrossRef]
- Deng, Y.; Yi, L.; Lin, X.; Lin, L.; Li, H.; Lin, J.M. A non-invasive genomic diagnostic method for bladder cancer using size-based filtration and microchip electrophoresis. Talanta 2015, 144, 136–144. [Google Scholar] [CrossRef]
- Planz, B.; Jochims, E.; Deix, T.; Caspers, H.P.; Jakse, G.; Boecking, A. The role of urinary cytology for detection of bladder cancer. Eur. J. Surg. Oncol. 2005, 31, 304–308. [Google Scholar] [CrossRef]
- Van Rhijn, B.W.; Van der Poel, H.G.; Van der Kwast, T.H. Urine markers for bladder cancer surveillance: A systematic review. Eur. Urol. 2005, 47, 736–748. [Google Scholar] [CrossRef]
- Tzouanas, C.; Lim, J.; Wen, Y.; Thiery, J.; Khoo, B. Microdevices for Non-Invasive Detection of Bladder Cancer. Chemosensors 2017, 5, 30. [Google Scholar] [CrossRef]
- Chahal, R.; Darshane, A.; Browning, A.J.; Sundaram, S.K. Evaluation of the clinical value of urinary NMP22 as a marker in the screening and surveillance of transitional cell carcinoma of the urinary bladder. Eur. Urol. 2001, 40, 415–420. [Google Scholar] [CrossRef]
- Guo, A.; Wang, X.; Gao, L.; Shi, J.; Sun, C.; Wan, Z. Bladder tumour antigen (BTA stat) test compared to the urine cytology in the diagnosis of bladder cancer: A meta-analysis. Can. Urol. Assoc. J. 2014, 8, E347–E352. [Google Scholar] [CrossRef]
- Brunzel, N.A. Fundamentals of Urine and Body Fluid Analysis; Elsevier: Amsterdam, The Netherlands, 2012; p. 464. [Google Scholar]
- Keshtkar, A.; Keshtkar, A.; Lawford, P. Cellular morphological parameters of the human urinary bladder (malignant and normal). Int. J. Exp. Pathol. 2007, 88, 185–190. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, D.; Sun, L.; Zhang, G.; Sun, L. Prognostic significance of the epithelial-to-mesenchymal transition markers e-cadherin, vimentin and twist in bladder cancer. Int. Braz. J. Urol. 2014, 40, 179–189. [Google Scholar] [CrossRef]
- Andersson, E.; Dahmcke, C.M.; Steven, K.; Larsen, L.K.; Guldberg, P. Filtration Device for On-Site Collection, Storage and Shipment of Cells from Urine and Its Application to DNA-Based Detection of Bladder Cancer. PLoS ONE 2015, 10, e0131889. [Google Scholar] [CrossRef]
- Birkhahn, M.; Mitra, A.P.; Williams, A.J.; Barr, N.J.; Skinner, E.C.; Stein, J.P.; Skinner, D.G.; Tai, Y.C.; Datar, R.H.; Cote, R.J. A novel precision-engineered microfiltration device for capture and characterisation of bladder cancer cells in urine. Eur. J. Cancer 2013, 49, 3159–3168. [Google Scholar] [CrossRef]
- Macgregor-Ramiasa, M.; McNicholas, K.; Ostrikov, K.; Li, J.; Michael, M.; Gleadle, J.M.; Vasilev, K. A platform for selective immuno-capture of cancer cells from urine. Biosens. Bioelectron. 2017, 96, 373–380. [Google Scholar] [CrossRef]
- Neoh, K.H.; Hassan, A.A.; Chen, A.; Sun, Y.; Liu, P.; Xu, K.F.; Wong, A.S.T.; Han, R.P.S. Rethinking liquid biopsy: Microfluidic assays for mobile tumor cells in human body fluids. Biomaterials 2018, 150, 112–124. [Google Scholar] [CrossRef]
- Chen, A.; Fu, G.; Xu, Z.; Sun, Y.; Chen, X.; Cheng, K.S.; Neoh, K.H.; Tang, Z.; Chen, S.; Liu, M.; et al. Detection of Urothelial Bladder Carcinoma via Microfluidic Immunoassay and Single-Cell DNA Copy-Number Alteration Analysis of Captured Urinary-Exfoliated Tumor Cells. Cancer Res. 2018, 78, 4073–4085. [Google Scholar] [CrossRef]
- Pichler, R.; Fritz, J.; Tulchiner, G.; Klinglmair, G.; Soleiman, A.; Horninger, W.; Klocker, H.; Heidegger, I. Increased accuracy of a novel mRNA-based urine test for bladder cancer surveillance. BJU Int. 2018, 121, 29–37. [Google Scholar] [CrossRef]
- Di Carlo, D.; Irimia, D.; Tompkins, R.G.; Toner, M. Continuous inertial focusing, ordering, and separation of particles in microchannels. Proc. Natl. Acad. Sci. USA 2007, 104, 18892–18897. [Google Scholar] [CrossRef]
- Parsa, S.; Gupta, M.; Loizeau, F.; Cheung, K.C. Effects of surfactant and gentle agitation on inkjet dispensing of living cells. Biofabrication 2010, 2, 025003. [Google Scholar] [CrossRef]
- Mustafi, D.; Smith, C.M.; Makinen, M.W.; Lee, R.C. Multi-block poloxamer surfactants suppress aggregation of denatured proteins. Biochim. Biophys. Acta 2008, 1780, 7–15. [Google Scholar] [CrossRef]
- Simerville, J.A.; Maxted, W.C.; Pahira, J.J. Urinalysis: A comprehensive review. Am. Fam. Physician 2005, 71, 1153–1162. [Google Scholar]
- Kuntaegowdanahalli, S.S.; Bhagat, A.A.; Kumar, G.; Papautsky, I. Inertial microfluidics for continuous particle separation in spiral microchannels. Lab Chip 2009, 9, 2973–2980. [Google Scholar] [CrossRef]
- Martel, J.M.; Toner, M. Inertial focusing dynamics in spiral microchannels. Phys. Fluids 2012, 24, 32001. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Singh, P.K.; Srivastava, K.; Singh, D.; Dalela, D.; Rath, S.K.; Goel, M.M.; Brahma Bhatt, M.L. Diagnostic role of survivin in urinary bladder cancer. Asian Pac. J. Cancer Prev. 2013, 14, 81–85. [Google Scholar] [CrossRef]
- Carlsson, J.; Wester, K.; De La Torre, M.; Malmstrom, P.U.; Gardmark, T. EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides. Radiol. Oncol. 2015, 49, 50–58. [Google Scholar] [CrossRef]
- Polioudaki, H.; Agelaki, S.; Chiotaki, R.; Politaki, E.; Mavroudis, D.; Matikas, A.; Georgoulias, V.; Theodoropoulos, P.A. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer 2015, 15, 399. [Google Scholar] [CrossRef]
- Acloque, H.; Thiery, J.P.; Nieto, M.A. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008, 9, 322–326. [Google Scholar] [CrossRef]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef]
- Myoteri, D.; Dellaportas, D.; Lykoudis, P.M.; Apostolopoulos, A.; Marinis, A.; Zizi-Sermpetzoglou, A. Prognostic Evaluation of Vimentin Expression in Correlation with Ki67 and CD44 in Surgically Resected Pancreatic Ductal Adenocarcinoma. Gastroenterol. Res. Pract. 2017, 2017, 9207616. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Shariat, S.F.; Palapattu, G.S.; Karakiewicz, P.I.; Rogers, C.G.; Vazina, A.; Bastian, P.J.; Schoenberg, M.P.; Lerner, S.P.; Sagalowsky, A.I.; Lotan, Y. Discrepancy between clinical and pathologic stage: Impact on prognosis after radical cystectomy. Eur. Urol. 2007, 51, 137–149. [Google Scholar] [CrossRef]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, Y.; Saya, H.; Arima, Y. Prospects for new lung cancer treatments that target EMT signaling. Dev. Dyn. 2018, 247, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.J.; Kim, W.J. Role of the epithelial-mesenchymal transition in bladder cancer: From prognosis to therapeutic target. Korean J. Urol. 2013, 54, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Rycaj, K. Targeted therapy against cancer stem cells. Oncol. Lett. 2015, 10, 27–33. [Google Scholar] [CrossRef]
- Guen, V.J.; Chavarria, T.E.; Kroger, C.; Ye, X.; Weinberg, R.A.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; Huang, T.; Wu, B.L.; He, W.X.; Liu, D. Stem cells in cancer therapy: Opportunities and challenges. Oncotarget 2017, 8, 75756–75766. [Google Scholar] [CrossRef]
- Nakai, Y.; Anai, S.; Kuwada, M.; Miyake, M.; Chihara, Y.; Tanaka, N.; Hirayama, A.; Yoshida, K.; Hirao, Y.; Fujimoto, K. Photodynamic diagnosis of shed prostate cancer cells in voided urine treated with 5-aminolevulinic acid. BMC Urol. 2014, 14, 59. [Google Scholar] [CrossRef]
- Vantaku, V.; Dong, J.; Ambati, C.R.; Perera, D.; Donepudi, S.R.; Amara, C.S.; Putluri, V.; Ravi, S.S.; Robertson, M.J.; Piyarathna, D.W.B.; et al. Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin. Cancer Res. 2019, 25, 3689–3701. [Google Scholar] [CrossRef]
- Amara, C.S.; Ambati, C.R.; Vantaku, V.; Piyarathna, D.W.; Donepudi, S.R.; Ravi, S.S.; Arnold, J.M.; Putluri, V.; Chatta, G.; Guru, K.A.; et al. Serum Metabolic Profiling Identified a Distinct Metabolic Signature in Bladder Cancer Smokers: A Key Metabolic Enzyme Associated with Patient Survival. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 770–781. [Google Scholar] [CrossRef]
- Warkiani, M.E.; Khoo, B.L.; Wu, L.; Tay, A.K.; Bhagat, A.A.; Han, J.; Lim, C.T. Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat. Protoc. 2016, 11, 134–148. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Age/Sex | Cytology | Staging | Metastasis | Diagnosis |
|---|---|---|---|---|---|
| 1 | 70/M | Inflammatory cells | cTaN0M0—Stage 0a | Nil | NMIBC |
| 2 | 69/M | NA | cT4N3M1 | Yes | MIBC—metastatic |
| 3 | 89/M | NA | cT3N0M0 | Nil | MIBC |
| 4 | 84/M | Atypical, reactive—negative for HG malignant cells | cTaN0M0—Stage 0a | Nil | NMIBC |
| 5 | 70/M | Highly atypical cells | cTaN0M0—Stage 0a | Nil | NMIBC |
| 6 | 74/M | HG malignant cells seen | cT2N0M0 | Nil | MIBC |
| 7 | 70/M | Highly atypical cells, consistent with urothelial Ca | cT3N0M0 | Nil | MIBC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoo, B.L.; Bouquerel, C.; Durai, P.; Anil, S.; Goh, B.; Wu, B.; Raman, L.; Mahendran, R.; Thamboo, T.; Chiong, E.; et al. Detection of Clinical Mesenchymal Cancer Cells from Bladder Wash Urine for Real-Time Detection and Prognosis. Cancers 2019, 11, 1274. https://doi.org/10.3390/cancers11091274
Khoo BL, Bouquerel C, Durai P, Anil S, Goh B, Wu B, Raman L, Mahendran R, Thamboo T, Chiong E, et al. Detection of Clinical Mesenchymal Cancer Cells from Bladder Wash Urine for Real-Time Detection and Prognosis. Cancers. 2019; 11(9):1274. https://doi.org/10.3390/cancers11091274
Chicago/Turabian StyleKhoo, Bee Luan, Charlotte Bouquerel, Pradeep Durai, Sarannya Anil, Benjamin Goh, Bingcheng Wu, Lata Raman, Ratha Mahendran, Thomas Thamboo, Edmund Chiong, and et al. 2019. "Detection of Clinical Mesenchymal Cancer Cells from Bladder Wash Urine for Real-Time Detection and Prognosis" Cancers 11, no. 9: 1274. https://doi.org/10.3390/cancers11091274
APA StyleKhoo, B. L., Bouquerel, C., Durai, P., Anil, S., Goh, B., Wu, B., Raman, L., Mahendran, R., Thamboo, T., Chiong, E., & Lim, C. T. (2019). Detection of Clinical Mesenchymal Cancer Cells from Bladder Wash Urine for Real-Time Detection and Prognosis. Cancers, 11(9), 1274. https://doi.org/10.3390/cancers11091274