High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study

 , , , , , , , , , and add
Show full author list
, , , , , , , , , and add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Patients’ Basic and Demographic Information
2.2. Expression Profiling of Tumor Samples
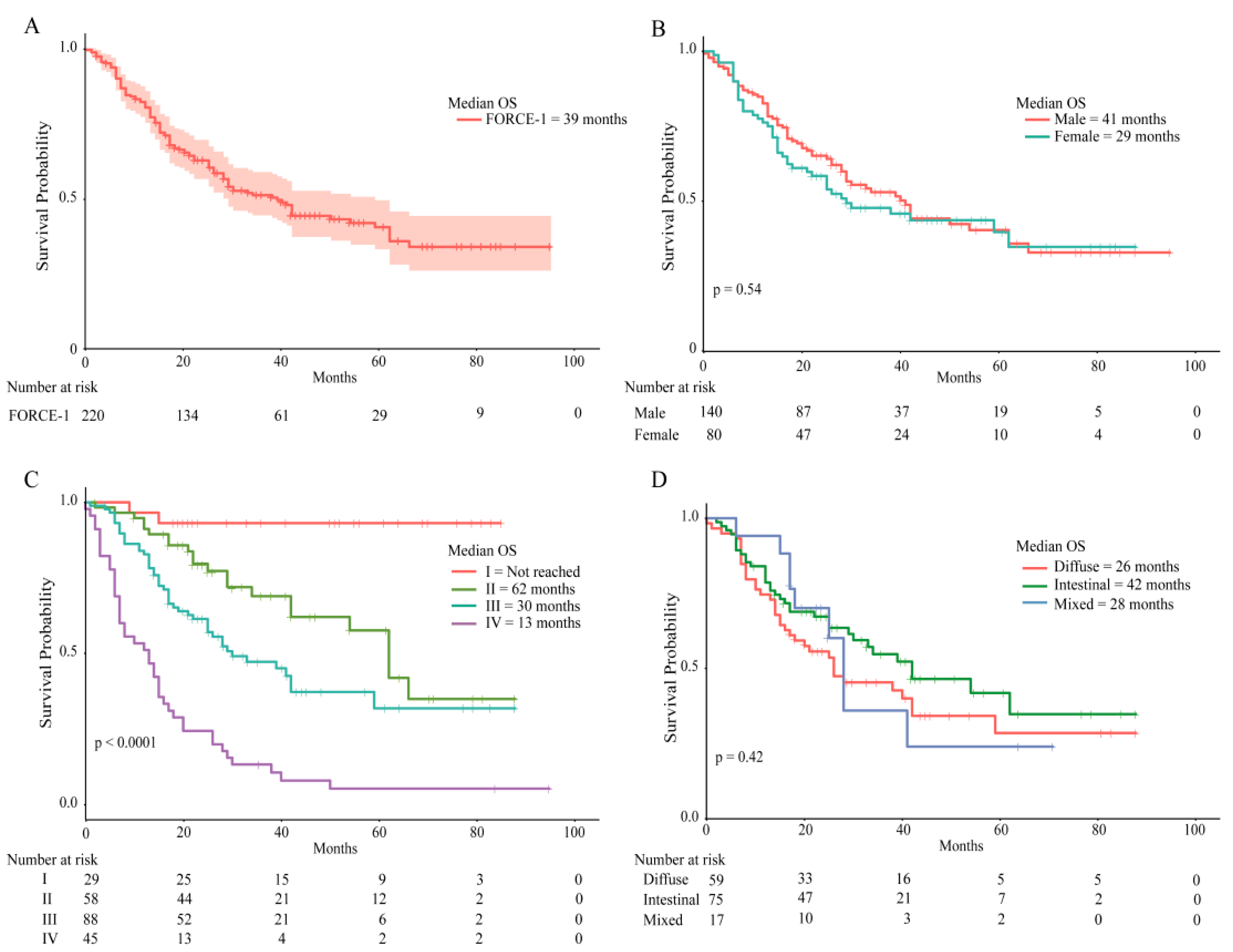
2.3. Patient Survival Rates
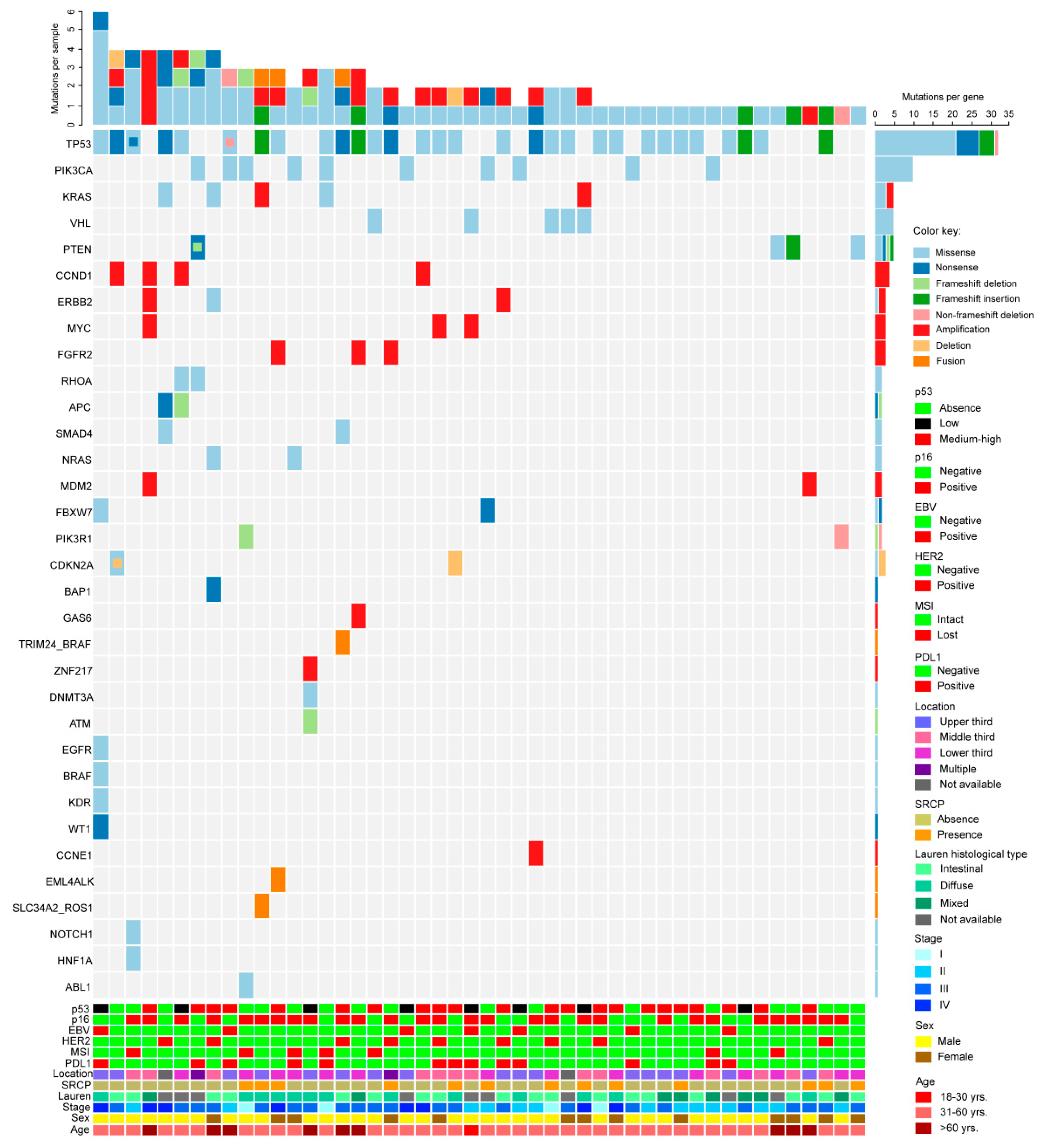
2.4. Next Generation Sequencing
2.5. Mutation Prevalence, Comparison with the TCGA Database
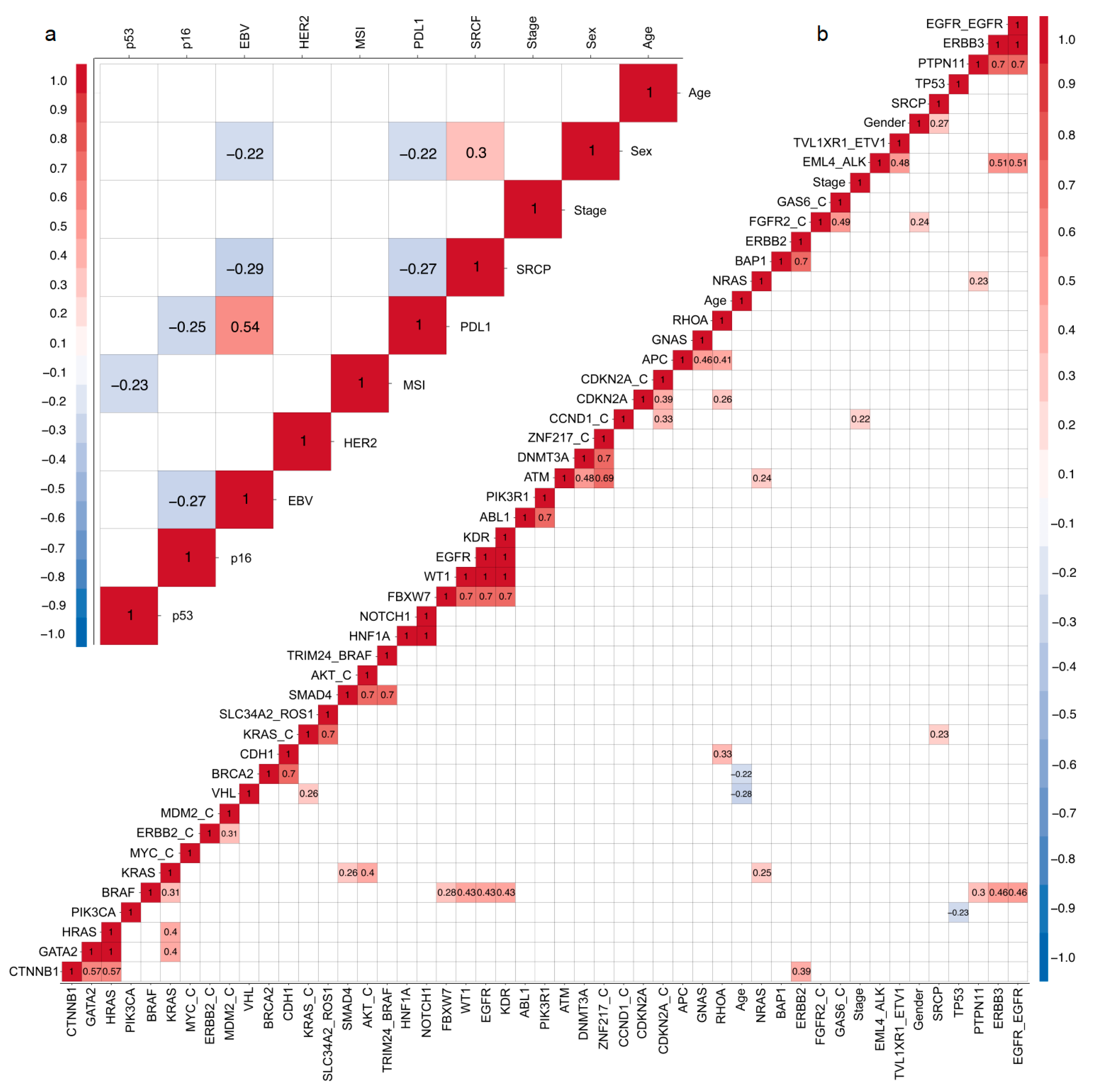
2.6. Correlation Matrix
3. Discussion
4. Materials and Methods
4.1. Patients, Ethics Approval, Consent to Publish and Demographic Data
4.2. Protein Expression Analyses by Tissue Microarray (TMA) and EBV Status
4.3. Next Generation Sequencing (NGS), DNA/RNA Purification & Quantification
4.4. Construction, Quantification, and Sequencing of Libraries
4.5. Bioinformatics Methods
4.6. Identification of Drivers and Druggable/Actionable Mutation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ruíz-García, E.; Guadarrama-Orozco, J.; Vidal-Millán, S.; Lino-Silva, L.S.; López-Camarillo, C.; Astudillo-de la Vega, H. Gastric cancer in Latin America. Scand. J. Gastroenterol. 2018, 53, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Jimenez De La Jara, J.; Bastias, G.; Ferreccio, C.; Moscoso, C.; Sagues, S.; Cid, C.; Bronstein, E.; Herrera, C.; Nervi, B.; Corvalan, A.; et al. A snapshot of cancer in Chile: Analytical frameworks for developing a cancer policy. Biol. Res. 2015, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours of the Digestive System, 4th ed.; Bosman, F.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; WHO: Lyon, France, 2010. [Google Scholar]
- Tan, I.B.; Ivanova, T.; Lim, K.H.; Ong, C.W.; Deng, N.; Lee, J.; Tan, S.H.; Wu, J.; Lee, M.H.; Ooi, C.H.; et al. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 2011, 141, 476–485.e11. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nature 2015, 21, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Caglevic, C.; Silva, S.; Mahave, M.; Rolfo, C.; Gallardo, J. The current situation for gastric cancer in Chile. Ecancermedicalscience 2016, 10, 8–15. [Google Scholar] [CrossRef][Green Version]
- Corvalan, A.; Koriyama, C.; Akiba, S.; Eizuru, Y.; Backhouse, C.; Palma, M.; Argandoña, J.; Tokunaga, M. Epstein-Barr virus in gastric carcinoma is associated with location in the cardia and with a diffuse histology: A study in one area of Chile. Int. J. Cancer 2001, 94, 527–530. [Google Scholar] [CrossRef]
- Corvalán, A.; Ruedlinger, J.; de Mayo, T.; Polakovicova, I.; Gonzalez-Hormazabal, P.; Aguayo, F. The Phylogeographic Diversity of EBV and Admixed Ancestry in the Americas–Another Model of Disrupted Human-Pathogen Co-Evolution. Cancers 2019, 11, 217. [Google Scholar] [CrossRef]
- Ferreccio, C.; Rollán, A.; Harris, P.R.; Serrano, C.; Gederlini, A.; Margozzini, P.; Gonzalez, C.; Aguilera, X.; Venegas, A.; Jara, A. Gastric cancer is related to early Helicobacter pylori infection in a high-prevalence country. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 662–667. [Google Scholar] [CrossRef]
- Carrasco-Avino, G.; Riquelme, I.; Padilla, O.; Villaseca, M.; Aguayo, F.R.; Corvalan, A.H. The Conundrum of the Epstein-Barr virus-associated gastric carcinoma in the Americas. Oncotarget 2017, 8, 75687–75698. [Google Scholar] [CrossRef] [PubMed]
- Owen, G.I.; Pinto, M.P.; Retamal, I.N.; Fernádez, M.F.; Cisternas, B.; Mondaca, S.; Sanchez, C.; Galindo, H.; Nervi, B.; Ibañez, C.; et al. Chilean Gastric Cancer Task Force: A study protocol to obtain a clinical and molecular classification of a cohort of gastric cancer patients. Medicine (Baltimore) 2018, 97, e0419. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Okada, E.; Ukawa, S.; Nakamura, K.; Hirata, M.; Nagai, A.; Matsuda, K.; Ninomiya, T.; Kiyohara, Y.; Muto, K.; Kamatani, Y.; et al. Demographic and lifestyle factors and survival among patients with esophageal and gastric cancer: The Biobank Japan Project. J. Epidemiol. 2017, 27, S29–S35. [Google Scholar] [CrossRef]
- Zong, L.; Abe, M.; Seto, Y.; Ji, J. The challenge of screening for early gastric cancer in China. Lancet 2016, 388, 2606. [Google Scholar] [CrossRef]
- Zhao, G.-H.; Li, T.-C.; Shi, L.-H.; Xia, Y.-B.; Lu, L.-M.; Huang, W.-B.; Sun, H.-L.; Zhang, Y.-S. Relationship between inactivation of p16 gene and gastric carcinoma. World J. Gastroenterol. 2003, 9, 905–909. [Google Scholar] [CrossRef]
- Giampieri, R.; Maccaroni, E.; Mandolesi, A.; Del Prete, M.; Andrikou, K.; Faloppi, L.; Bittoni, A.; Bianconi, M.; Scarpelli, M.; Bracci, R.; et al. Mismatch repair deficiency may affect clinical outcome through immune response activation in metastatic gastric cancer patients receiving first-line chemotherapy. Gastric Cancer 2017, 20, 156–163. [Google Scholar] [CrossRef]
- Shah, M.A.; Khanin, R.; Tang, L.; Janjigian, Y.Y.; Klimstra, D.S.; Gerdes, H.; Kelsen, D.P. Molecular classification of gastric cancer: A new paradigm. Clin. Cancer Res. 2011, 17, 2693–2701. [Google Scholar] [CrossRef]
- Ichikawa, H.; Nagahashi, M.; Shimada, Y.; Hanyu, T.; Ishikawa, T.; Kameyama, H.; Kobayashi, T.; Sakata, J.; Yabusaki, H.; Nakagawa, S.; et al. Actionable gene-based classification toward precision medicine in gastric cancer. Genome Med. 2017, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Sher, X.; Park, S.H.; Kang, W.K.; Jung, H.; Lee, M.; Loboda, A.; Kim, S.T.; Lee, J.; Kang, P.S.; Lee, S.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar]
- Mishima, S.; Kuwata, T.; Yoshino, T.; Doi, T.; Kawazoe, A.; Tsuji, A.; Nakamura, Y.; Kojima, T.; Shitara, K.; Ohtsu, A.; et al. Clinicopathological and molecular features of responders to nivolumab for patients with advanced gastric cancer. J. Immunother. Cancer 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Urakami, K.; Mochizuki, T.; Serizawa, M.; Shimoda, Y.; Yamaguchi, K.; Kusuhara, M.; Naruoka, A.; Akiyama, Y.; Ohnami, S.; et al. Tumor mutational burden analysis of 2,000 Japanese cancer genomes using whole exome and targeted gene panel sequencing. Biomed. Res. 2018, 39, 159–167. [Google Scholar]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Zhuang, W.; Ma, J.; Chen, X.; Wang, G.; Lu, J.; Chen, Y.; Dong, H.; Cai, S.; Zhang, Y.; Zhao, X.; et al. The tumor mutational Burden of Chinese advanced cancer patients estimated by a 381-cancer-gene Panel. J. Cancer 2018, 9, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Shibata, T.; Okamoto, Y.; Yamazaki, J.; Kawamura, T.; Horiguchi, N.; Okubo, M.; Nakano, N.; Ishizuka, T.; Nagasaka, M.; et al. Mutation spectrum of TP53 gene predicts clinicopathological features and survival of gastric cancer. Oncotarget 2016, 7, 42252–42260. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio-Preiser, C.M.; Wang, J.; Stemmermann, G.N.; Noffsinger, A. TP53 and gastric carcinoma: A review. Hum. Mutat. 2003, 21, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kang, M.S.; Kim, K.M. Epstein-Barr virus-associated gastric carcinoma and specific features of the accompanying immune response. J. Gastric Cancer 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Constanza Camargo, M.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.Y.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, J.; Qiu, H.; Kong, P.; Chen, S.; Li, W.; Zhan, Y.; Li, Y.; Chen, Y.; Zhou, Z.; et al. Prognostic significance of Epstein-Barr virus infection in gastric cancer: A meta-analysis. BMC Cancer 2015, 15, 782. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Kang, B.W.; Kwon, O.K.; Park, K.B.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Jeon, S.W.; Kang, H.; et al. Intratumoural PD-l1 expression is associated with worse survival of patients with epstein– barr virus-associated gastric cancer. Br. J. Cancer 2017, 117, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Kankeu Fonkoua, L.; Yee, N. Molecular Characterization of Gastric Carcinoma: Therapeutic Implications for Biomarkers and Targets. Biomedicines 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kim, S.; Lai, J.F.; Hyung, W.J.; Choi, W.H.; Choi, S.H.; Noh, S.H. Advanced gastric carcinoma with signet ring cell histology. Oncology 2007, 72, 64–68. [Google Scholar] [CrossRef]
- Whitehall, V.L.J.; Buttenshaw, R.L.; Bond, C.E.; Montgomery, G.W.; Ramsnes, I.; Greco, S.A.; Zhen Zhao, Z.; Leggett, B.A.; Umapathy, A.; Mallitt, K.-A. p53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int. J. Cancer 2011, 130, 1567–1576. [Google Scholar]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Chau, I.; Baselga, J.; Tabernero, J.; Elez-Fernandez, M.E.; Sirzen, F.; Wolf, J.; Hidalgo, M.; Gervais, R.; Blay, J.-Y.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Chiurillo, M.A. Role of the Wnt/β-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Tan, G.A.; Giri, D.D.; Zhou, X.K.; Howe, L.R. Activation status of Wnt/ß-catenin signaling in normal and neoplastic breast tissues: Relationship to HER2/neu expression in human and mouse. PLoS ONE 2012, 7, e33421. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, G.; Diaz, J.; Valbuena, J.R.; Ibanez, P.; Rodriguez, P.; Araya, G.; Rodriguez, C.; Torres, J.; Duarte, I.; Aravena, E.; et al. Overexpression of p73 as a Tissue Marker for High-Risk Gastritis. Clin. Cancer Res. 2010, 16, 3253–3259. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Vargas, M.; Ossandón, F.; Santibáñez, E.; Urrutia, J.; Luengo, V.; Zavala, L.F.; Backhouse, C.; Palma, M.; Argandoña, J.; et al. DNA methylation profile in diffuse type gastric cancer: Evidence for hypermethylation of the BRCA1 promoter region in early-onset gastric carcinogenesis. Biol. Res. 2008, 41, 303–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kulangara, K.; Hanks, D.A.; Waldroup, S.; Peltz, L.; Shah, S.; Roach, C.; Juco, J.W.; Emancipator, K.; Stanforth, D. Development of the combined positive score (CPS) for the evaluation of PD-L1 in solid tumors with the immunohistochemistry assay PD-L1 IHC 22C3 pharmDx. J. Clin. Oncol. 2017, 35, e14589. [Google Scholar] [CrossRef]
- Rüschoff, J.; Hanna, W.; Bilous, M.; Hofmann, M.; Osamura, R.Y.; Penault-Llorca, F.; Van De Vijver, M.; Viale, G. HER2 testing in gastric cancer: A practical approach. Mod. Pathol. 2012, 25, 637–650. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef]
- Voss, M.H.; Snyder, A.; Zhang, H.; Hantash, F.M.; Berger, M.F.; Riely, G.J.; Chi, P.; Callahan, M.K.; Sabbatini, P.; Lowery, M.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | FORCE-1 n (%) | TMA n (%) | NGS n (%) |
|---|---|---|---|
| Gender | |||
| Male | 142 (63.4) | 58 (64.4) | 67 (66.3) |
| Female | 82 (36.6) | 32 (35.6) | 34 (33.7) |
| Stage at diagnosis | |||
| I | 30 (13.4) | 8 (8.9) | 9 (8.9) |
| II | 57 (25.4) | 24 (26.7) | 31 (30.7) |
| III | 88 (39.3) | 46 (51.1) | 49 (48.5) |
| IV | 49 (21.9) | 12 (13.3) | 12 (11.9) |
| ECOG Performance Status | |||
| 0 | 69 (30.8) | 25 (27.8) | 29 (28.7) |
| 1 | 69 (30.8) | 27 (30.0) | 26 (25.7) |
| 2 | 6 (2.7) | 2 (2.2) | 3 (3.0) |
| ≥3 | 1 (0.4) | 1 (1.1) | 1 (1.0) |
| NA | 79 (35.3) | 35 (38.8) | 42 (41.6) |
| Location of primary tumor | |||
| Distal esophagus and GEJ | 49 (21.9) | 18 (20.0) | 24 (23.8) |
| Fundus | 12 (5.3) | 8 (8.9) | 8 (7.9) |
| Corpus | 86 (38.4) | 28 (31.1) | 32 (31.7) |
| Antrum | 54 (24.1) | 23 (25.6) | 25 (24.8) |
| Pylorus | 8 (3.6) | 5 (5.6) | 6 (5.9) |
| Multiple | 9 (4.0) | 4 (4.4) | 3 (3.0) |
| NA | 6 (2.7) | 4 (4.4) | 3 (3.0) |
| Lauren histological type | |||
| Intestinal | 76 (33.9) | 27 (30.0) | 34 (33.7) |
| Diffuse | 61 (27.2) | 32 (35.6) | 30 (29.7) |
| Mixed | 17 (7.6) | 13 (14.4) | 14 (13.9) |
| NA | 70 (31.3) | 18 (20.0) | 23 (22.8) |
| WHO histological type | |||
| Adenocarcinoma | 171 (76.3) | 71 (78.9) | 80 (79.2) |
| Undifferentiated carcinoma | 9 (4.0) | 6 (6.7) | 4 (4.0) |
| Adenosquamous cell carcinoma | 3 (1.3) | 3 (3.3) | 3 (3.0) |
| NA | 41 (18.3) | 10 (11.1) | 14 (13.9) |
| Signet-ring cell presence | |||
| No | 122 (54.5) | 32 (35.6) | 59 (58.4) |
| Yes | 74 (33.0) | 53 (58.9) | 34 (33.7) |
| NA | 28 (12.5) | 5 (5.6) | 8 (7.9) |
| Comorbidities at diagnosis | |||
| Two or less | 192 (85.7) | 79 (87.8) | 84 (83.2) |
| Three or more | 32 (14.3) | 11 (12.2) | 17 (16.7) |
| Age | |||
| Mean, median (range) | 61.4, 62 (26–89) | 62.7, 63 (26–89) | 60.9, 62 (27–88) |
| IHC Analysis | n (%) |
|---|---|
| PDL-1 a | |
| Negative | 64 (71.1) |
| Positive | 26 (28.9) |
| MSI+ (MMR deficient) | 13 (14.4) |
| MLH-1 | |
| Intact | 78 (86.7) |
| Lost | 12 (13.3) |
| PMS-2 | |
| Intact | 78 (86.7) |
| Lost | 12 (13.3) |
| MSH-2 | |
| Intact | 90 (100) |
| Lost | 0 |
| MSH-6 | |
| Intact | 88 (97.8) |
| Lost | 2 (2.2) |
| HER-2 | |
| Negative | 78 (86.7) |
| Positive | 12 (13.3) |
| CISH-EBV | |
| Negative | 78 (86.7) |
| Positive | 12 (13.3) |
| p16 | |
| Absence | 33 (36.7) |
| Presence | 57 (63.3) |
| p53 | |
| Intact | 42 (46.7) |
| Mutated | 48 (53.3) |
| Mutation Gene | Frequency n (%) | Mutation Gene | Frequency n (%) |
|---|---|---|---|
| SNVs | CNVs | ||
| TP53 | 49 (48.51) | MYC a | 5 (4.95) |
| PIK3CA | 15 (14.85) | CCND1 a | 4 (3.96) |
| VHL | 6 (5.94) | CCNE a | 4 (3.96) |
| NRAS | 7 (6.93) | FGFR2 a | 4 (3.96) |
| KRAS | 6 (5.94) | ERBB2 a | 3 (2.97) |
| BRAF | 5 (4.95) | MDM2 a | 3 (2.97) |
| APC | 5 (4.95) | CDKN2A b | 2 (1.98) |
| PTEN | 5 (4.95) | KRAS a | 2 (1.98) |
| RHOA | 4 (3.96) | AKT1 a | 1 (0.99) |
| CDKN2A | 3 (2.97) | CDK6 a | 1 (0.99) |
| CTNNB1 | 3 (2.97) | GAS6 a | 1 (0.99) |
| ATM | 2 (1.98) | ZNF217 a | 1 (0.99) |
| PIK3R1 | 2 (1.98) | ||
| PTPN11 | 2 (1.98) | Fusions | |
| ERBB3 | 1 (0.99) | EML4_ALK | 4 (4.65) |
| FBXW7 | 2 (1.98) | EGFR_EGFR | 1 (1.16) |
| DNMT3A | 2 (1.98) | SLC34A2_ROS1 | 1 (1.16) |
| SMAD4 | 2 (1.98) | TBL1XR1_ETV1 | 1 (1.16) |
| CDH1 | 2 (1.98) | TRIM24_BRAF | 1 (1.16) |
| ERBB2 | 2 (1.98) | ||
| Gene | Aminoacidic Mutational Change | Total Samples n | 101 | 100 | 30 | 295 | 66 |
|---|---|---|---|---|---|---|---|
| Function | Chilean FORCE-1 n (%) | UHK TCGA n (%) | UTOKIO TCGA n (%) | TCGA Nature 2014 n (%) | Brazil TCGA n (%) | ||
| TP53 | R273C | Missense | 4 (4.0%) | 4 (4.0%) | NR | 6 (2.0%) | 1 (1.5%) |
| R213 * | Nonsense | 3 (3.0%) | 1 (1.0%) | NR | 5 (1.7%) | NR | |
| R175H | Missense | 2 (2.0%) | NR | 1 (3.3%) | 6 (2.0%) | 1 (1.5%) | |
| R248Q | Missense | 2 (2.0%) | 4 (4.0%) | NR | 5 (1.7%) | 3 (4.5%) | |
| R248W | Missense | 2 (2.0%) | 1 (1.0%) | NR | 1 (0.3%) | NR | |
| P98S | Missense | 2 (2.0%) | NR | NR | NR | NR | |
| Y220H | Missense | 2 (2.0%) | NR | NR | NR | 2 (3%) | |
| C242F | Missense | 2 (2.0%) | NR | NR | NR | NR | |
| PIK3CA | E542K | Missense | 4 (4.0%) | NR | NR | 5 (1.7%) | 2 (3%) |
| C378R | Missense | 2 (2.0%) | NR | NR | 1 (0.3%) | NR | |
| E545K | Missense | 2 (2.0%) | NR | NR | 11 (3.7%) | 2 (3%) | |
| R88Q | Missense | 2 (2.0%) | NR | NR | 4 (1.4%) | 1 (1.5%) | |
| T1025A | Missense | 2 (2.0%) | NR | NR | NR | NR | |
| VHL | S68L | Missense | 6 (5.9%) | NR | NR | NR | NR |
| NRAS | G13V | Missense | 5 (5.0%) | NR | NR | NR | NR |
| G12D | Missense | 2 (2.0%) | NR | NR | NR | NR | |
| KRAS | G12D | Missense | 3 (3.0%) | 2 (2%) | NR | 7 (2.4%) | 1 (1.5%) |
| BRAF | D594G | Missense | 3 (3.0%) | NR | NR | NR | NR |
| RHOA | Y42C | Missense | 3 (3.0%) | NR | 4 (13.3%) | 3 (1%) | NR |
| APC | D156fs | Frameshift deletion | 2 (2.0%) | NR | NR | NR | NR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordova-Delgado, M.; Pinto, M.P.; Retamal, I.N.; Muñoz-Medel, M.; Bravo, M.L.; Fernández, M.F.; Cisternas, B.; Mondaca, S.; Sanchez, C.; Galindo, H.; et al. High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study. Cancers 2019, 11, 1275. https://doi.org/10.3390/cancers11091275
Cordova-Delgado M, Pinto MP, Retamal IN, Muñoz-Medel M, Bravo ML, Fernández MF, Cisternas B, Mondaca S, Sanchez C, Galindo H, et al. High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study. Cancers. 2019; 11(9):1275. https://doi.org/10.3390/cancers11091275
Chicago/Turabian StyleCordova-Delgado, Miguel, Mauricio P. Pinto, Ignacio N. Retamal, Matías Muñoz-Medel, María Loreto Bravo, María F. Fernández, Betzabé Cisternas, Sebastián Mondaca, César Sanchez, Hector Galindo, and et al. 2019. "High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study" Cancers 11, no. 9: 1275. https://doi.org/10.3390/cancers11091275
APA StyleCordova-Delgado, M., Pinto, M. P., Retamal, I. N., Muñoz-Medel, M., Bravo, M. L., Fernández, M. F., Cisternas, B., Mondaca, S., Sanchez, C., Galindo, H., Nervi, B., Ibáñez, C., Acevedo, F., Madrid, J., Peña, J., Koch, E., Maturana, M. J., Romero, D., de la Jara, N., ... Garrido, M. (2019). High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study. Cancers, 11(9), 1275. https://doi.org/10.3390/cancers11091275