Baseline Genomic Features in BRAFV600-Mutated Metastatic Melanoma Patients Treated with BRAF Inhibitor + MEK Inhibitor in Routine Care

 ,
,  ,
, 
 ,
, 

Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Baseline DNA Analysis (Mutations and CNVs)
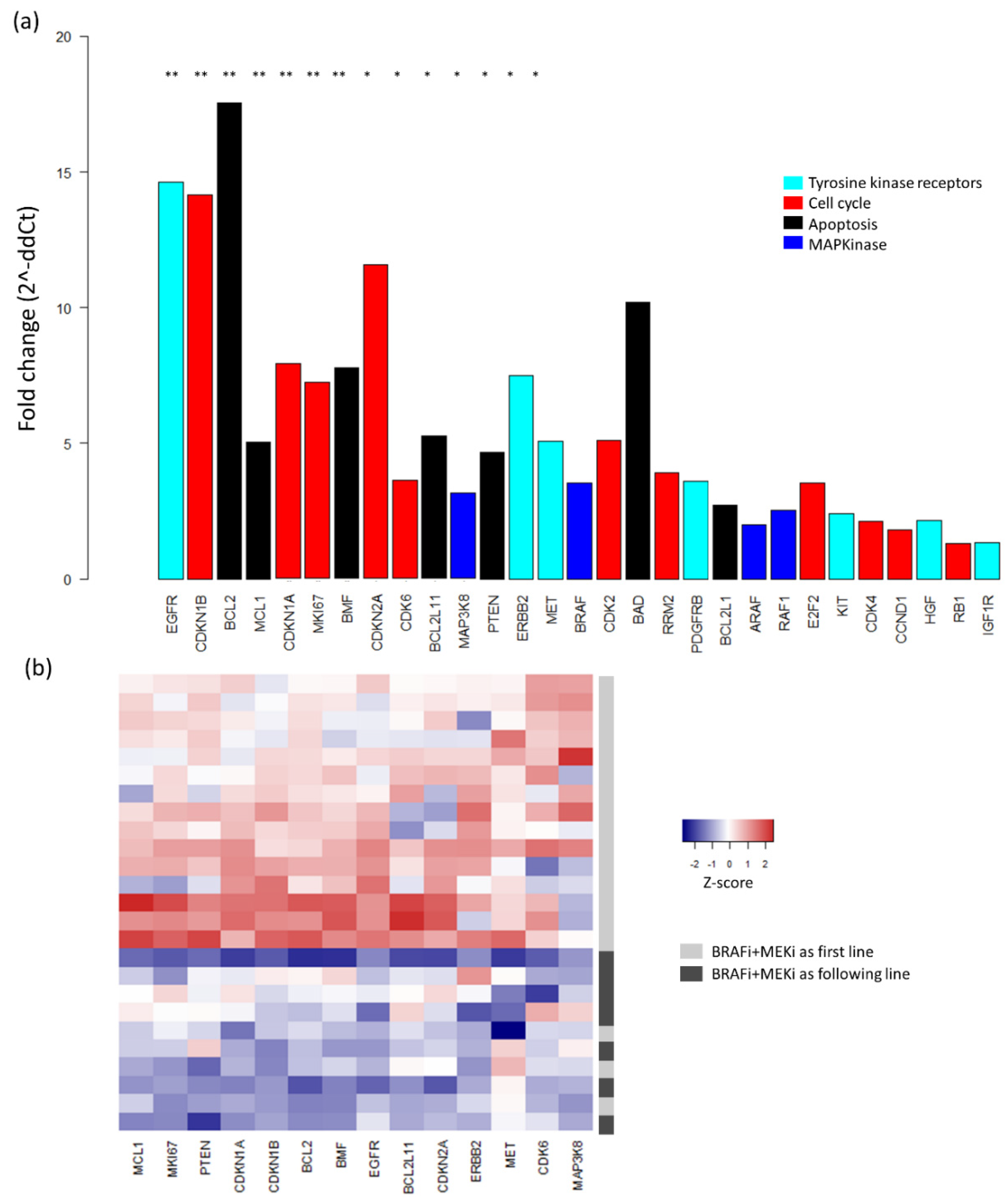
2.3. Baseline mRNA Expression Analysis
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. DNA and mRNA Extractions
4.3. Copy Number Variation and Transcript Analyses
4.4. Next-Generation Sequencing (NGS)
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Manzano, J.L.; Layos, L.; Bugés, C.; de los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Alicea, G.M.; Paraiso, K.H.T.; Lawrence, H.; Gibney, G.T.; Smalley, K.S.M. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014, 27, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gopal, Y.N.V.; Scott, A.; Chen, G.; Woodman, S.E.; Davies, M.A. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res. 2012, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.-K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Louveau, B.; Delyon, J.; De Moura, C.R.; Battistella, M.; Jouenne, F.; Golmard, L.; Sadoux, A.; Podgorniak, M.-P.; Chami, I.; Marco, O.; et al. A targeted genomic alteration analysis predicts survival of melanoma patients under BRAF inhibitors. Oncotarget 2019, 10, 1669–1687. [Google Scholar] [CrossRef]
- Wongchenko, M.J.; McArthur, G.A.; Dréno, B.; Larkin, J.; Ascierto, P.A.; Sosman, J.; Andries, L.; Kockx, M.; Hurst, S.D.; Caro, I.; et al. Gene Expression Profiling in BRAF-Mutated Melanoma Reveals Patient Subgroups with Poor Outcomes to Vemurafenib That May Be Overcome by Cobimetinib Plus Vemurafenib. Clin. Cancer Res. 2017, 23, 5238–5245. [Google Scholar] [CrossRef]
- Yan, Y.; Wongchenko, M.J.; Robert, C.; Larkin, J.; Ascierto, P.A.; Dréno, B.; Maio, M.; Garbe, C.; Chapman, P.B.; Sosman, J.A.; et al. Genomic Features of Exceptional Response in Vemurafenib ± Cobimetinib-treated Patients with BRAFV600-mutated Metastatic Melanoma. Clin. Cancer Res. 2019, 25, 3239–3246. [Google Scholar] [CrossRef]
- Wagle, M.-C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M.; et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018, 2, 7. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Larkin, J.M.G.; Yan, Y.; McArthur, G.A.; Ascierto, P.A.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.V.; Stroyakovskiy, D.; Thomas, L.; et al. Update of progression-free survival (PFS) and correlative biomarker analysis from coBRIM: Phase III study of cobimetinib (cobi) plus vemurafenib (vem) in advanced BRAF-mutated melanoma. JCO 2015, 33, 9006. [Google Scholar] [CrossRef]
- Cavalieri, S.; Di Guardo, L.; Cimminiello, C.; Bono, A.; Tolomio, E.; Colombetti, A.; Valeri, B.; Di Tolla, G.; de Braud, F.; Del Vecchio, M. Combined therapy with dabrafenib and trametinib in BRAF-mutated metastatic melanoma in a real-life setting: The INT Milan experience. Tumori 2016, 102, 501–507. [Google Scholar] [CrossRef]
- Depenni, R.; De Rosa, F.; Greco, S.; Ridolfi, L.; Pellacani, G.; Ponti, G.; Cascinu, S.; Guidoboni, M. Dabrafenib-trametinib combination in “field-practice”: An Italian experience. Future Oncol. 2018, 14, 2045–2052. [Google Scholar] [CrossRef]
- Luke, J.J.; Ghate, S.R.; Kish, J.; Lee, C.H.; McAllister, L.; Mehta, S.; Ndife, B.; Feinberg, B.A. Targeted agents or immuno-oncology therapies as first-line therapy for BRAF-mutated metastatic melanoma: A real-world study. Future Oncol. 2019. [Google Scholar] [CrossRef]
- Carlino, M.S.; Gowrishankar, K.; Saunders, C.A.B.; Pupo, G.M.; Snoyman, S.; Zhang, X.D.; Saw, R.; Becker, T.M.; Kefford, R.F.; Long, G.V.; et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol. Cancer Ther. 2013, 12, 1332–1342. [Google Scholar] [CrossRef]
- Flaherty, K.; Davies, M.A.; Grob, J.J.; Long, G.V.; Nathan, P.; Ribas, A.; Robert, C. Genomic analysis and 3-year efficacy and safety update of COMBI-d. J. Clin. Oncol. 2019, 34, 9502. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Kunz, M.; Hölzel, M. The impact of melanoma genetics on treatment response and resistance in clinical and experimental studies. Cancer Metastasis Rev. 2017, 36, 53–75. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Neiswender, J.V.; Kortum, R.L.; Bourque, C.; Kasheta, M.; Zon, L.I.; Morrison, D.K.; Ceol, C.J. KIT Suppresses BRAFV600E-Mutant Melanoma by Attenuating Oncogenic RAS/MAPK Signaling. Cancer Res. 2017, 77, 5820–5830. [Google Scholar] [CrossRef]
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF inhibition causes resilience of melanoma cell lines by inducing the secretion of FGF1. Oncogenesis 2018, 7, 71. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Dvinge, H.; Bertone, P. HTqPCR: High-throughput analysis and visualization of quantitative real-time PCR data in R. Bioinformatics 2009, 25, 3325–3326. [Google Scholar] [CrossRef]
- Lebbé, C.; How-Kit, A.; Battistella, M.; Sadoux, A.; Podgorniak, M.-P.; Sidina, I.; Pages, C.; Roux, J.; Porcher, R.; Tost, J.; et al. BRAF(V600) mutation levels predict response to vemurafenib in metastatic melanoma. Melanoma Res. 2014, 24, 415–418. [Google Scholar] [CrossRef]
- Chraybi, M.; Abd Alsamad, I.; Copie-Bergman, C.; Baia, M.; André, J.; Dumaz, N.; Ortonne, N. Oncogene abnormalities in a series of primary melanomas of the sinonasal tract: NRAS mutations and cyclin D1 amplification are more frequent than KIT or BRAF mutations. Hum. Pathol. 2013, 44, 1902–1911. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Included Patients | |
|---|---|
| (n = 27) | |
| Male sex | 13 (48%) |
| Age at melanoma diagnosis, years | 49.3 (35.5; 58.8) |
| Melanoma subtype | |
| Superficial spreading melanoma | 15 (56%) |
| Nodular melanoma | 5 (19%) |
| Others | 2 (7%) |
| Undetermined | 5 (19%) |
| Breslow thickness, mm | 3 (1.4; 5.2) |
| Ulceration | 11/24 (46%) |
| Age at initiation, years | 49.9 (40.8; 62.7) |
| Disease stage | |
| Unresectable stage III | 3 (11%) |
| Unresectable stage IV | 24 (89%) |
| M1c | 16 (67%) |
| Brain metastases | 6 (22%) |
| Elevated lactate dehydrogenase level | 7/24 (29%) |
| Number of disease sites | |
| ≤2 | 13/24 (54%) |
| ≥3 | 11/24 (46%) |
| BRAFi + MEKi combination initiated | |
| Dabrafenib + trametinib | 24 (89%) |
| Vemurafenib + cobimetinib | 3 (11%) |
| BRAFi + MEKi as first-line treatment | |
| First-line treatment | 20 (74%) |
| Following-line treatment | 7 (26%) |
| Previous line of treatment (if any) | |
| BRAFi monotherapy | 3 (11%) |
| Anti-CTLA4 | 2 (7%) |
| Anti-PD1/PDL1 | 6 (22%) |
| Anti-CTLA4 + anti-PD1 | 2 (7%) |
| Others | 1 (4%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louveau, B.; Jouenne, F.; Reger de Moura, C.; Sadoux, A.; Baroudjian, B.; Delyon, J.; Herms, F.; De Masson, A.; Da Meda, L.; Battistella, M.; et al. Baseline Genomic Features in BRAFV600-Mutated Metastatic Melanoma Patients Treated with BRAF Inhibitor + MEK Inhibitor in Routine Care. Cancers 2019, 11, 1203. https://doi.org/10.3390/cancers11081203
Louveau B, Jouenne F, Reger de Moura C, Sadoux A, Baroudjian B, Delyon J, Herms F, De Masson A, Da Meda L, Battistella M, et al. Baseline Genomic Features in BRAFV600-Mutated Metastatic Melanoma Patients Treated with BRAF Inhibitor + MEK Inhibitor in Routine Care. Cancers. 2019; 11(8):1203. https://doi.org/10.3390/cancers11081203
Chicago/Turabian StyleLouveau, Baptiste, Fanelie Jouenne, Coralie Reger de Moura, Aurelie Sadoux, Barouyr Baroudjian, Julie Delyon, Florian Herms, Adele De Masson, Laetitia Da Meda, Maxime Battistella, and et al. 2019. "Baseline Genomic Features in BRAFV600-Mutated Metastatic Melanoma Patients Treated with BRAF Inhibitor + MEK Inhibitor in Routine Care" Cancers 11, no. 8: 1203. https://doi.org/10.3390/cancers11081203
APA StyleLouveau, B., Jouenne, F., Reger de Moura, C., Sadoux, A., Baroudjian, B., Delyon, J., Herms, F., De Masson, A., Da Meda, L., Battistella, M., Dumaz, N., Lebbe, C., & Mourah, S. (2019). Baseline Genomic Features in BRAFV600-Mutated Metastatic Melanoma Patients Treated with BRAF Inhibitor + MEK Inhibitor in Routine Care. Cancers, 11(8), 1203. https://doi.org/10.3390/cancers11081203