NF-κB Signaling in Ovarian Cancer

Abstract
1. Introduction
2. Canonical and Non-Canonical NF-κB Signaling in OC
2.1. Canonical NF-κB Signaling in Proliferation and Chemoresistance
2.2. Non-Canonical NF-κB Signaling and OC Persistence
2.3. NF-κB in Promoting Stemness
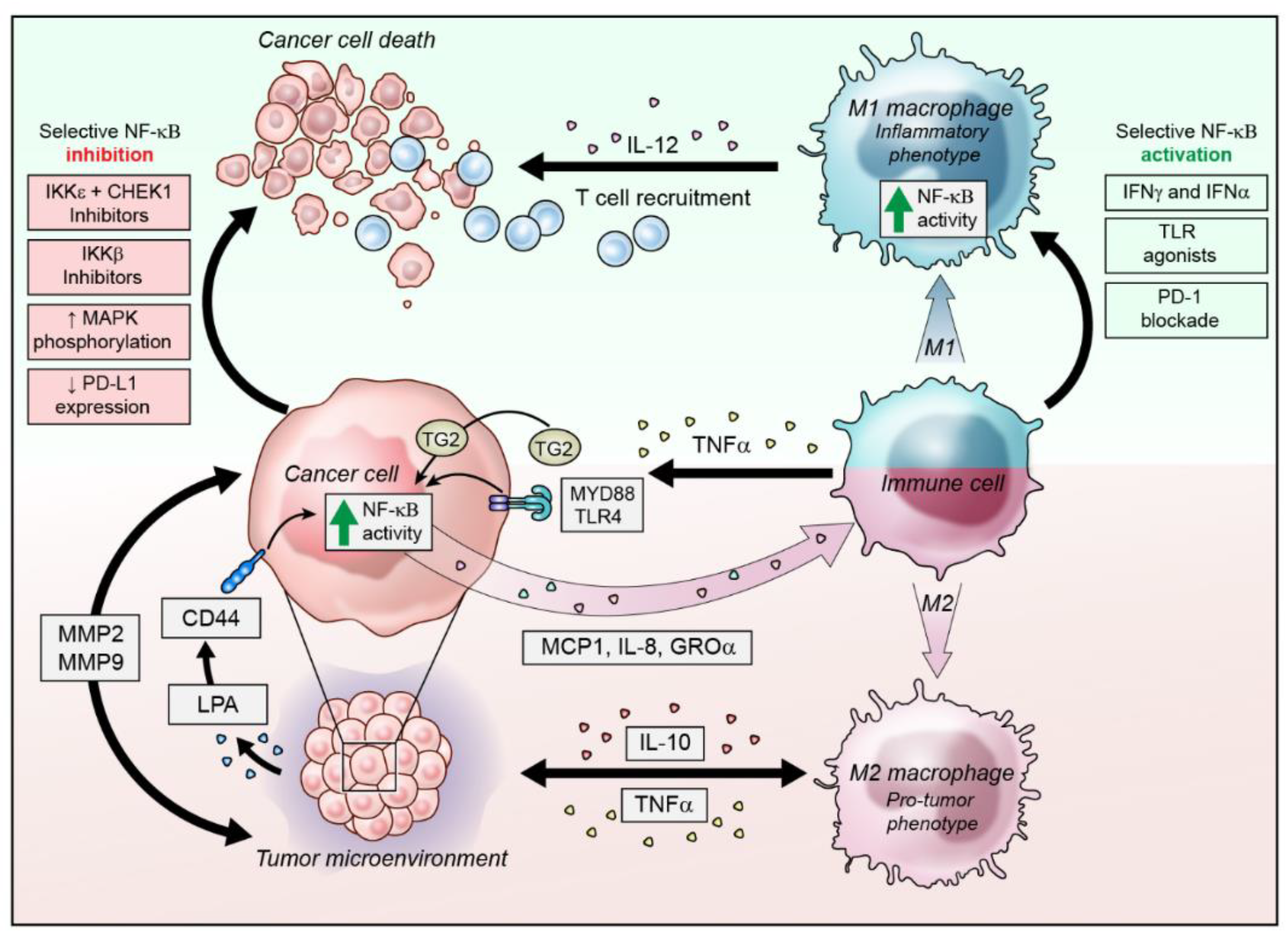
3. NF-κB Activity in the Inflammatory Ovarian Cancer Microenvironment
3.1. Modulation of NF-κB Activity in Infiltrating Immune Cells
3.2. NF-κB Activation in Ovarian Cancer Cells to Alter the Microenvironment
4. Strategies to Target NF-κB in Ovarian Cancer
4.1. IKK Inhibition
4.2. Toll-Like Receptors and Myeloid Differentiation Primary Response 88 (Myd-88)
4.3. Lysophosphatidic Acid (LPA)
4.4. microRNAs
4.5. Targeted NF-κB Activation for Anti-Tumor Immunity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Kamal, R.; Hamed, S.; Mansour, S.; Mounir, Y.; Abdel Sallam, S. Ovarian cancer screening-ultrasound; impact on ovarian cancer mortality. Br. J. Radiol. 2018, 91, 20170571. [Google Scholar] [CrossRef] [PubMed]
- House, C.D.; Hernandez, L.; Annunziata, C.M. In vitro enrichment of ovarian cancer tumor-initiating cells. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- House, C.D.; Jordan, E.; Hernandez, L.; Ozaki, M.; James, J.M.; Kim, M.; Kruhlak, M.J.; Batchelor, E.; Elloumi, F.; Cam, M.C.; et al. NFkappaB Promotes Ovarian Tumorigenesis via Classical Pathways That Support Proliferative Cancer Cells and Alternative Pathways That Support ALDH(+) Cancer Stem-like Cells. Cancer Res. 2017, 77, 6927–6940. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front. Oncol. 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Mitrakas, L.; Gravas, S.; Papandreou, C.; Koukoulis, G.; Karasavvidou, F.; Dimakopoulos, G.; Weingartner, K.; Karatzas, A.; Zachos, I.; Tzortzis, V. Primary High-Grade Non-Muscle-Invasive Bladder Cancer: High NFkappaB Expression in Tumor Specimens Distinguishes Patients Who are at Risk for Disease Progression. Pathol. Oncol. Res. 2019, 25, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, J.; Li, N.; Sun, N.; Wang, C. Expression of nuclear factor-kappaB and its clinical significance in nonsmall-cell lung cancer. Ann. Thorac. Surg. 2006, 82, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L.; Yap, N.Y.; Rajandram, R.; Small, D.; Pailoor, J.; Ong, T.A.; Razack, A.H.; Wood, S.T.; Morais, C.; Gobe, G.C. Nuclear factor-kappa B subunits and their prognostic cancer-specific survival value in renal cell carcinoma patients. Pathology 2018, 50, 511–518. [Google Scholar] [CrossRef]
- Lua, J.; Qayyum, T.; Edwards, J.; Roseweir, A.K. The Prognostic Role of the Non-Canonical Nuclear Factor-Kappa B Pathway in Renal Cell Carcinoma Patients. Urol. Int. 2018, 101, 190–196. [Google Scholar] [CrossRef]
- Plewka, D.; Kowalczyk, A.E.; Jakubiec-Bartnik, B.; Morek, M.; Bogunia, E.; Kmiec, A.; Wierzbicki, P.M.; Plewka, A. Immunohistochemical visualization of pro-inflammatory cytokines and enzymes in ovarian tumors. Folia Histochem. Cytobiol. 2014, 52, 124–137. [Google Scholar] [CrossRef][Green Version]
- Giopanou, I.; Bravou, V.; Papanastasopoulos, P.; Lilis, I.; Aroukatos, P.; Papachristou, D.; Kounelis, S.; Papadaki, H. Metadherin, p50, and p65 expression in epithelial ovarian neoplasms: an immunohistochemical study. Biomed. Res. Int. 2014, 2014, 178410. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Stavnes, H.T.; Kleinberg, L.; Berner, A.; Hernandez, L.F.; Birrer, M.J.; Steinberg, S.M.; Davidson, B.; Kohn, E.C. Nuclear factor kappaB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 2010, 116, 3276–3284. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Hsu, S.C.; Davidson, B.; Birrer, M.J.; Kohn, E.C.; Annunziata, C.M. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res. 2010, 70, 4005–4014. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Kim, M.; Hernandez, L.; Grajales, V.; Noonan, A.; Anver, M.; Davidson, B.; Annunziata, C.M. IKK-epsilon coordinates invasion and metastasis of ovarian cancer. Cancer Res. 2012, 72, 5494–5504. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.P.; Shu, S.K.; He, L.; Lee, Y.C.; Kruk, P.A.; Grenman, S.; Nicosia, S.V.; Mor, G.; Schell, M.J.; Coppola, D.; et al. Deregulation of IKBKE is associated with tumor progression, poor prognosis, and cisplatin resistance in ovarian cancer. Am. J. Pathol. 2009, 175, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Fuller, P.J. Characterization of the inhibitor of kappaB kinase (IKK) complex in granulosa cell tumors of the ovary and granulosa cell tumor-derived cell lines. Horm. Cancer 2013, 4, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Block, M.S.; Vierkant, R.A.; Fogarty, Z.C.; Winham, S.J.; Visscher, D.W.; Kalli, K.R.; Wang, C.; Goode, E.L. The inflammatory microenvironment in epithelial ovarian cancer: A role for TLR4 and MyD88 and related proteins. Tumor Biol. 2016, 37, 13279–13286. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, C.; Jin, S.; Qu, D.; Ying, H. Expression of NF-kappaB and PTEN in primary epithelial ovarian carcinoma and the correlation with chemoresistance. Int. J. Clin. Exp. Pathol. 2015, 8, 10953–10963. [Google Scholar] [PubMed]
- Darb-Esfahani, S.; Sinn, B.V.; Weichert, W.; Budczies, J.; Lehmann, A.; Noske, A.; Buckendahl, A.C.; Muller, B.M.; Sehouli, J.; Koensgen, D.; et al. Expression of classical NF-kappaB pathway effectors in human ovarian carcinoma. Histopathology 2010, 56, 727–739. [Google Scholar] [CrossRef]
- Gaikwad, S.M.; Thakur, B.; Sakpal, A.; Singh, R.K.; Ray, P. Differential activation of NF-kappaB signaling is associated with platinum and taxane resistance in MyD88 deficient epithelial ovarian cancer cells. Int. J. Biochem. Cell Biol. 2015, 61, 90–102. [Google Scholar] [CrossRef]
- Momeny, M.; Yousefi, H.; Eyvani, H.; Moghaddaskho, F.; Salehi, A.; Esmaeili, F.; Alishahi, Z.; Barghi, F.; Vaezijoze, S.; Shamsaiegahkani, S.; et al. Blockade of nuclear factor-kappaB (NF-kappaB) pathway inhibits growth and induces apoptosis in chemoresistant ovarian carcinoma cells. Int. J. Biochem. Cell Biol. 2018, 99, 9. [Google Scholar] [CrossRef] [PubMed]
- Shuang, T.; Wang, M.; Zhou, Y.; Shi, C. Over-expression of nuclear NF-kappaB1 and c-Rel correlates with chemoresistance and prognosis of serous epithelial ovarian cancer. Exp. Mol. Pathol. 2016, 100, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.K.; Huang, S.L.; Chang, T.C.; Chao, C.C. TLR4 and NFkappaB signaling is critical for taxol resistance in ovarian carcinoma cells. J. Cell. Physiol. 2018, 233, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xiao, X.; Rosen, D.G.; Cheng, X.; Wu, X.; Chang, B.; Liu, G.; Xue, F.; Mercado-Uribe, I.; Chiao, P.; et al. The biphasic role of NF-kappaB in progression and chemoresistance of ovarian cancer. Clin. Cancer Res. 2011, 17, 2181–2194. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Torres, C.; Gaytan-Cervantes, J.; Vazquez-Santillan, K.; Mandujano-Tinoco, E.A.; Ceballos-Cancino, G.; Garcia-Venzor, A.; Zampedri, C.; Sanchez-Maldonado, P.; Mojica-Espinosa, R.; Jimenez-Hernandez, L.E.; et al. NF-kappaB Participates in the Stem Cell Phenotype of Ovarian Cancer Cells. Arch. Med. Res. 2017, 48, 343–351. [Google Scholar] [CrossRef]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Forster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A Role for NF-kappaB in Organ Specific Cancer and Cancer Stem Cells. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular Pathways: The Balance between Cancer and the Immune System Challenges the Therapeutic Specificity of Targeting Nuclear Factor-kappaB Signaling for Cancer Treatment. Clin. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef]
- Struzik, J.; Szulc-Dabrowska, L. NF-kappaB Signaling in Targeting Tumor Cells by Oncolytic Viruses-Therapeutic Perspectives. Cancers 2018, 10. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.K.; Huang, H.J.; Le, X.F.; Mao, W.; Lu, K.H.; Bast, R.C., Jr.; Liao, W.S. MEKK3 expression correlates with nuclear factor kappa B activity and with expression of antiapoptotic genes in serous ovarian carcinoma. Cancer 2009, 115, 3897–3908. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Shabeeb, D.; Musa, A.E. NF-kappaB targeting for overcoming tumor resistance and normal tissues toxicity. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Singha, B.; Gatla, H.R.; Phyo, S.; Patel, A.; Chen, Z.S.; Vancurova, I. IKK inhibition increases bortezomib effectiveness in ovarian cancer. Oncotarget 2015, 6, 26347–26358. [Google Scholar] [CrossRef]
- Huang, S.; Robinson, J.B.; Deguzman, A.; Bucana, C.D.; Fidler, I.J. Blockade of nuclear factor-kappaB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res. 2000, 60, 5334–5339. [Google Scholar]
- Wilson, A.J.; Barham, W.; Saskowski, J.; Tikhomirov, O.; Chen, L.; Lee, H.J.; Yull, F.; Khabele, D. Tracking NF-kappaB activity in tumor cells during ovarian cancer progression in a syngeneic mouse model. J. Ovarian Res. 2013, 6, 63. [Google Scholar] [CrossRef]
- Hong, L.; Wang, S.; Li, W.; Wu, D.; Chen, W. Tumor-associated macrophages promote the metastasis of ovarian carcinoma cells by enhancing CXCL16/CXCR6 expression. Pathol. Res. Pract. 2018, 214, 1345–1351. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef]
- Kleinschmidt, E.G.; Miller, N.L.G.; Ozmadenci, D.; Tancioni, I.; Osterman, C.D.; Barrie, A.M.; Taylor, K.N.; Ye, A.; Jiang, S.; Connolly, D.C.; et al. Rgnef promotes ovarian tumor progression and confers protection from oxidative stress. Oncogene 2019. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Sedukhina, A.S.; Okamoto, N.; Nagasawa, S.; Suzuki, N.; Ohta, T.; Hattori, H.; Roche-Molina, M.; Narvaez, A.J.; Jeyasekharan, A.D.; et al. NF-kappaB signaling mediates acquired resistance after PARP inhibition. Oncotarget 2015, 6, 3825–3839. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Ma, X.; Hua, W.; Chen, B.; Cai, G. Caveolin-1 mediates chemoresistance in cisplatin-resistant ovarian cancer cells by targeting apoptosis through the Notch-1/Akt/NF-kappaB pathway. Oncol. Rep. 2015, 34, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Koti, M.; Gooding, R.J.; Nuin, P.; Haslehurst, A.; Crane, C.; Weberpals, J.; Childs, T.; Bryson, P.; Dharsee, M.; Evans, K.; et al. Identification of the IGF1/PI3K/NF kappaB/ERK gene signalling networks associated with chemotherapy resistance and treatment response in high-grade serous epithelial ovarian cancer. BMC Cancer 2013, 13, 549. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, B.K.; Ding, X.; Coombe, K.; Baldwin, A.S.; Kim, H.J. Active roles for inhibitory kappaB kinases alpha and beta in nuclear factor-kappaB-mediated chemoresistance to doxorubicin. Mol. Cancer Ther. 2008, 7, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Witty, J.M.; Rocha, S.; Perkins, N.D. Cisplatin mimics ARF tumor suppressor regulation of RelA (p65) nuclear factor-kappaB transactivation. Cancer Res. 2006, 66, 929–935. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- Roy, L.; Cowden Dahl, K.D. Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Sorrentino, C.; Ucar, D.A.; Peng, Y.; Matossian, M.; Wyczechowska, D.; Crabtree, J.; Zabaleta, J.; Morello, S.; Del Valle, L.; et al. Notch Signaling Regulates Mitochondrial Metabolism and NF-kappaB Activity in Triple-Negative Breast Cancer Cells via IKKalpha-Dependent Non-canonical Pathways. Front. Oncol. 2018, 8, 575. [Google Scholar] [CrossRef]
- Margalef, P.; Colomer, C.; Villanueva, A.; Montagut, C.; Iglesias, M.; Bellosillo, B.; Salazar, R.; Martinez-Iniesta, M.; Bigas, A.; Espinosa, L. BRAF-induced tumorigenesis is IKKalpha-dependent but NF-kappaB-independent. Sci. Signal 2015, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Yakubov, B.; Chelladurai, B.; Schmitt, J.; Emerson, R.; Turchi, J.J.; Matei, D. Extracellular tissue transglutaminase activates noncanonical NF-kappaB signaling and promotes metastasis in ovarian cancer. Neoplasia 2013, 15, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Cao, L.; Shen, C.; Satpathy, M.; Chelladurai, B.; Bigsby, R.M.; Nakshatri, H.; Matei, D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.L.; Liu, S.H.; Ai, Z.H.; Tao, M.F.; Ma, L.; Wen, S.Y.; Dai, M.; Liu, F.; Liu, H.S.; Jiang, R.Z.; et al. RelB/NF-kappaB links cell cycle transition and apoptosis to endometrioid adenocarcinoma tumorigenesis. Cell Death Dis. 2016, 7, e2402. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Chefetz, I.; Alvero, A.B.; Holmberg, J.C.; Lebowitz, N.; Craveiro, V.; Yang-Hartwich, Y.; Yin, G.; Squillace, L.; Gurrea Soteras, M.; Aldo, P.; et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 2013, 12, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Guzman, M.E.; Ibanez Hernandez, M.; Gomez-Gallegos, A.A.; Ortiz-Sanchez, E. ALDH as a Stem Cell marker in solid tumors. Curr. Stem Cell Res. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Cho, U.; Kim, B.; Kim, S.; Han, Y.; Song, Y.S. Pro-inflammatory M1 macrophage enhances metastatic potential of ovarian cancer cells through NF-kappaB activation. Mol. Carcinog. 2018, 57, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Karyampudi, L.; Lamichhane, P.; Krempski, J.; Kalli, K.R.; Behrens, M.D.; Vargas, D.M.; Hartmann, L.C.; Janco, J.M.; Dong, H.; Hedin, K.E.; et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-kappaB. Cancer Res. 2016, 76, 239–250. [Google Scholar] [CrossRef]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Kelly, M.G.; Fest, S.; Visintin, I.; Leiser, A.; Schwartz, P.E.; Rutherford, T.; Mor, G. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 2008, 27, 4712–4723. [Google Scholar] [CrossRef]
- Nishio, H.; Yaguchi, T.; Sugiyama, J.; Sumimoto, H.; Umezawa, K.; Iwata, T.; Susumu, N.; Fujii, T.; Kawamura, N.; Kobayashi, A.; et al. Immunosuppression through constitutively activated NF-kappaB signalling in human ovarian cancer and its reversal by an NF-kappaB inhibitor. Br. J. Cancer 2014, 110, 2965–2974. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, G.; Huang, J.; Ma, S.; Mi, K.; Cheng, J.; Zhu, Y.; Zha, X.; Huang, W. Atractylenolide I modulates ovarian cancer cell-mediated immunosuppression by blocking MD-2/TLR4 complex-mediated MyD88/NF-kappaB signaling in vitro. J. Transl. Med. 2016, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Yang, M.; Ding, Y.; Chen, J. Microbial infection, inflammation and epithelial ovarian cancer. Oncol. Lett. 2017, 14, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Mascellino, M.T.; Boccia, P.; Oliva, A. Immunopathogenesis in Chlamydia trachomatis Infected Women. ISRN Obstet. Gynecol. 2011, 2011, 436936. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-kappaB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen 2016, 21, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-kappaB Kinase (IKK) alpha and Nuclear Factor-kappaB (NFkappaB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7. [Google Scholar] [CrossRef]
- Prescott, J.A.; Cook, S.J. Targeting IKKbeta in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKbeta Inhibitors. Cells 2018, 7. [Google Scholar] [CrossRef]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef]
- Kinose, Y.; Sawada, K.; Makino, H.; Ogura, T.; Mizuno, T.; Suzuki, N.; Fujikawa, T.; Morii, E.; Nakamura, K.; Sawada, I.; et al. IKKbeta Regulates VEGF Expression and Is a Potential Therapeutic Target for Ovarian Cancer as an Antiangiogenic Treatment. Mol. Cancer Ther. 2015, 14, 909–919. [Google Scholar] [CrossRef]
- Sawada, I.; Hashimoto, K.; Sawada, K.; Kinose, Y.; Nakamura, K.; Toda, A.; Nakatsuka, E.; Yoshimura, A.; Mabuchi, S.; Fujikawa, T.; et al. The Novel IkappaB Kinase beta Inhibitor, IMD-0560, Has Potent Therapeutic Efficacy in Ovarian Cancer Xenograft Model Mice. Int. J. Gynecol. Cancer 2016, 26, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Min, D.J.; Wright, G.; Goldlust, I.; Annunziata, C.M. Loss of compensatory pro-survival and anti-apoptotic modulator, IKKepsilon, sensitizes ovarian cancer cells to CHEK1 loss through an increased level of p21. Oncotarget 2014, 5, 12788–12802. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Ogawa, M.; Muto, S.; Itai, A.; Isobe, M.; Hirata, Y.; Nagai, R. Novel IkB kinase inhibitors for treatment of nuclear factor-kB-related diseases. Expert Opin. Investig. Drugs 2011, 20, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Shifera, A.S. Protein-protein interactions involving IKKgamma (NEMO) that promote the activation of NF-kappaB. J. Cell. Physiol. 2010, 223, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.A.; Dougherty, P.G.; Cooper, J.K.; Qian, Z.; Lindert, S.; Wang, Q.E.; Pei, D. Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IkappaB Kinase Interaction Directly from a Combinatorial Library. J. Am. Chem. Soc. 2018, 140, 12102–12110. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Reilly, S.M.; Gomez, A.V.; Meral, R.; Butz, L.; Ajluni, N.; Chenevert, T.L.; Korytnaya, E.; Neidert, A.H.; Hench, R.; et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 157–170.e157. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Kim, K.H.; Jo, M.S.; Suh, D.S.; Yoon, M.S.; Shin, D.H.; Lee, J.H.; Choi, K.U. Expression and significance of the TLR4/MyD88 signaling pathway in ovarian epithelial cancers. World J. Surg. Oncol. 2012, 10, 193. [Google Scholar] [CrossRef]
- Block, M.S.; Vierkant, R.A.; Rambau, P.F.; Winham, S.J.; Wagner, P.; Traficante, N.; Toloczko, A.; Tiezzi, D.G.; Taran, F.A.; Sinn, P.; et al. MyD88 and TLR4 Expression in Epithelial Ovarian Cancer. Mayo Clin. Proc. 2018, 93, 307–320. [Google Scholar] [CrossRef]
- Wang, A.C.; Ma, Y.B.; Wu, F.X.; Ma, Z.F.; Liu, N.F.; Gao, R.; Gao, Y.S.; Sheng, X.G. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma in vitro. Oncol. Lett. 2014, 7, 871–877. [Google Scholar] [CrossRef]
- Zandi, Z.; Kashani, B.; Poursani, E.M.; Bashash, D.; Kabuli, M.; Momeny, M.; Mousavi-Pak, S.H.; Sheikhsaran, F.; Alimoghaddam, K.; Mousavi, S.A.; et al. TLR4 blockade using TAK-242 suppresses ovarian and breast cancer cells invasion through the inhibition of extracellular matrix degradation and epithelial-mesenchymal transition. Eur. J. Pharmacol. 2019, 853, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Zhang, G.N.; Shi, Y.; Zha, X.; Zhu, Y.; Wang, M.M.; Lin, Q.; Wang, W.; Lu, H.Y.; Ma, S.Q.; et al. Atractylenolide-I sensitizes human ovarian cancer cells to paclitaxel by blocking activation of TLR4/MyD88-dependent pathway. Sci. Rep. 2014, 4, 3840. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Chuffa, L.G.; de Moura Ferreira, G.; Lupi, L.A.; da Silva Nunes, I.; Favaro, W.J. P-MAPA immunotherapy potentiates the effect of cisplatin on serous ovarian carcinoma through targeting TLR4 signaling. J. Ovarian Res. 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Green, D.S.; Nunes, A.T.; Annunziata, C.M.; Zoon, K.C. Monocyte and interferon based therapy for the treatment of ovarian cancer. Cytokine Growth Factor Rev. 2016, 29, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Wang, F.Q.; Wu, H.S.; Mukherjee, T.J.; Fishman, D.A. The NF-kappaB pathway mediates lysophosphatidic acid (LPA)-induced VEGF signaling and cell invasion in epithelial ovarian cancer (EOC). Gynecol. Oncol. 2011, 123, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.M.; Pu, Y.; Han, Z.; Liu, T.; Li, Y.X.; Liu, M.; Li, X.; Tang, H. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. FEBS J. 2009, 276, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Teng, Y.; Yang, H.; Ma, J. Propofol inhibits invasion and growth of ovarian cancer cells via regulating miR-9/NF-kappaB signal. Braz. J. Med. Biol. Res. 2016, 49, e5717. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Tang, W.; Lin, Z.; Xu, L.; Dong, R.; Li, Y.; Li, J.; Zhang, Z.; Li, X.; et al. miR-130a upregulates mTOR pathway by targeting TSC1 and is transactivated by NF-kappaB in high-grade serous ovarian carcinoma. Cell Death Differ. 2017, 24, 2089–2100. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. MicroRNA-503-5p Inhibits the CD97-Mediated JAK2/STAT3 Pathway in Metastatic or Paclitaxel-Resistant Ovarian Cancer Cells. Neoplasia 2019, 21, 206–215. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, X.L.; Bai, R.; Liu, W.Y.; Li, X.; Liu, M.; Tang, H. miR-23a promotes IKKalpha expression but suppresses ST7L expression to contribute to the malignancy of epithelial ovarian cancer cells. Br. J. Cancer 2016, 115, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Shuang, T.; Wang, M.; Zhou, Y.; Shi, C.; Wang, D. NF-kappaB1, c-Rel and ELK1 inhibit miR-134 expression leading to TAB1 upregulation in paclitaxel-resistant human ovarian cancer. Oncotarget 2017, 8, 24853–24868. [Google Scholar] [CrossRef] [PubMed]
- van Jaarsveld, M.T.; Helleman, J.; Boersma, A.W.; van Kuijk, P.F.; van Ijcken, W.F.; Despierre, E.; Vergote, I.; Mathijssen, R.H.; Berns, E.M.; Verweij, J.; et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene 2013, 32, 4284–4293. [Google Scholar] [CrossRef] [PubMed]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24. [Google Scholar] [CrossRef]
- Ortega, R.A.; Barham, W.; Sharman, K.; Tikhomirov, O.; Giorgio, T.D.; Yull, F.E. Manipulating the NF-kappaB pathway in macrophages using mannosylated, siRNA-delivering nanoparticles can induce immunostimulatory and tumor cytotoxic functions. Int. J. Nanomed. 2016, 11, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Chieppa, M.; Bianchi, G.; Solinas, G.; Fabbri, M.; Laskarin, G.; Mantovani, A. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin. Dev. Immunol. 2010, 2010, 547179. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Facciabene, A.; Brady, W.E.; Aghajanian, C.A.; Fracasso, P.M.; Walker, J.L.; Lankes, H.A.; Manjarrez, K.L.; Danet-Desnoyers, G.H.; Bell-McGuinn, K.M.; et al. Integrative Development of a TLR8 Agonist for Ovarian Cancer Chemoimmunotherapy. Clin. Cancer Res. 2017, 23, 1955–1966. [Google Scholar] [CrossRef]
- Monk, B.J.; Brady, M.F.; Aghajanian, C.; Lankes, H.A.; Rizack, T.; Leach, J.; Fowler, J.M.; Higgins, R.; Hanjani, P.; Morgan, M.; et al. A phase 2, randomized, double-blind, placebo- controlled study of chemo-immunotherapy combination using motolimod with pegylated liposomal doxorubicin in recurrent or persistent ovarian cancer: A Gynecologic Oncology Group partners study. Ann. Oncol. 2017, 28, 996–1004. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| microRNA | Function Related to NF-κB Activity | References |
|---|---|---|
| miR-9 | miR-9 targeted the 3’UTR of NF-κB to suppress NF-κB activation and MMP-9 expression leading to reduced invasion and cell growth. | [97,98] |
| miR-130α | miR-130a was activated by NF-κB signaling, miR-130a promoted proliferation and invasion of OC cells. | [99] |
| miR-503-5p | NF-κB-downregulated miR-503-5p and activated the JAK/STAT pathway. Inhibition of miR-503-5p by NF-κB enhanced invasion and migration in vitro. | [100] |
| miR-23α | miR-23a increased the expression of IKKα which promoted growth, migration, and invasion in OC cells in vitro | [101] |
| miR-134 | TAB1 promoted pro-survival and anti-apoptosis via NF-κB signaling, TAB1 was inhibited by miR-134. NF-κB binds to the putative promoter region of miR-134 and represses its expression. | [102] |
| miR-141 | miR-141 downregulates KEAP1 and activates NF-κB to mediate cisplatin resistance in vitro | [103] |
| miR-199a | miR-199a negatively regulates IKKβ expression in OC cells. | [69] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrington, B.S.; Annunziata, C.M. NF-κB Signaling in Ovarian Cancer. Cancers 2019, 11, 1182. https://doi.org/10.3390/cancers11081182
Harrington BS, Annunziata CM. NF-κB Signaling in Ovarian Cancer. Cancers. 2019; 11(8):1182. https://doi.org/10.3390/cancers11081182
Chicago/Turabian StyleHarrington, Brittney S., and Christina M. Annunziata. 2019. "NF-κB Signaling in Ovarian Cancer" Cancers 11, no. 8: 1182. https://doi.org/10.3390/cancers11081182
APA StyleHarrington, B. S., & Annunziata, C. M. (2019). NF-κB Signaling in Ovarian Cancer. Cancers, 11(8), 1182. https://doi.org/10.3390/cancers11081182