Papillary Thyroid Carcinoma Variants are Characterized by Co-dysregulation of Immune and Cancer Associated Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
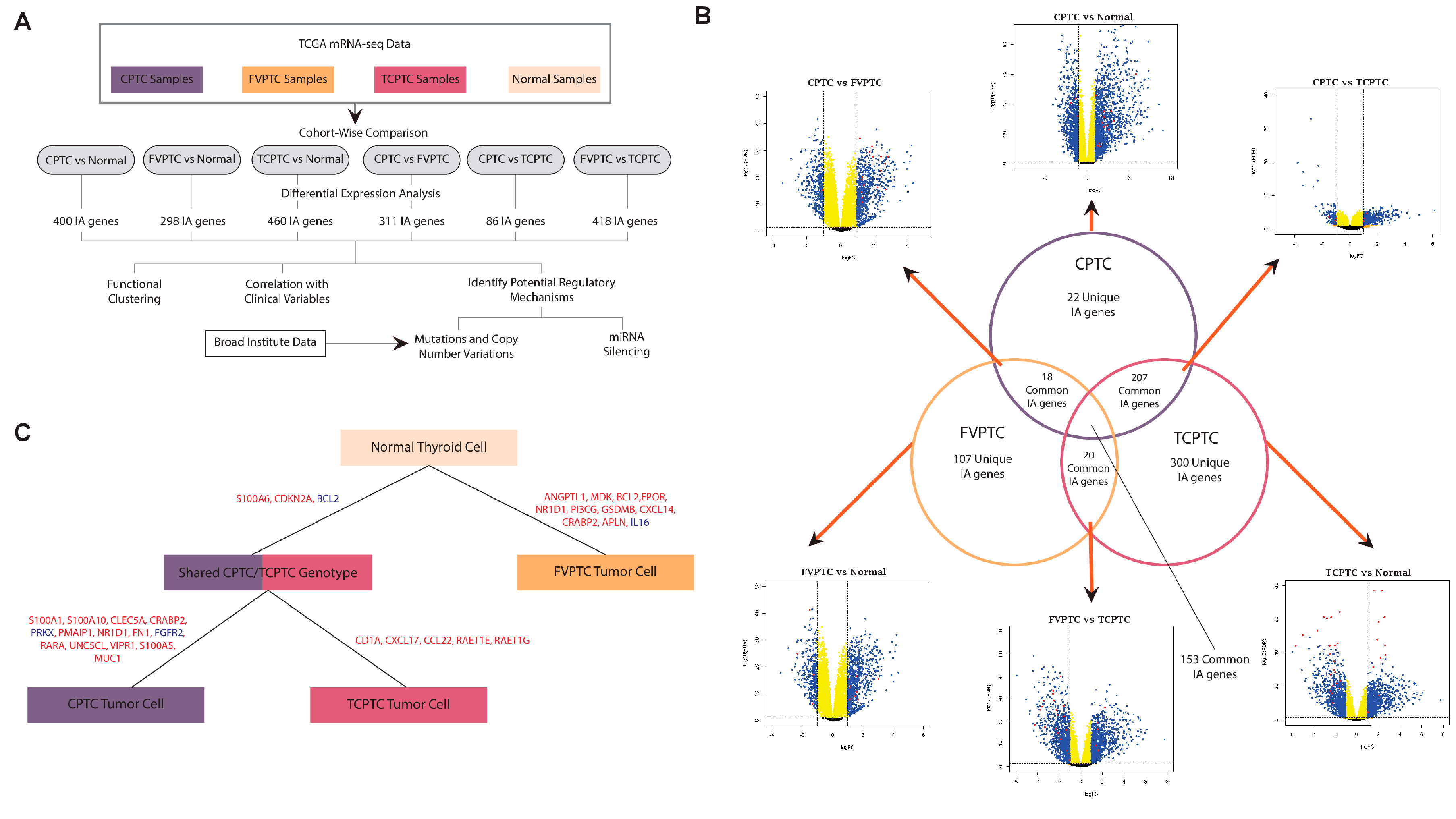
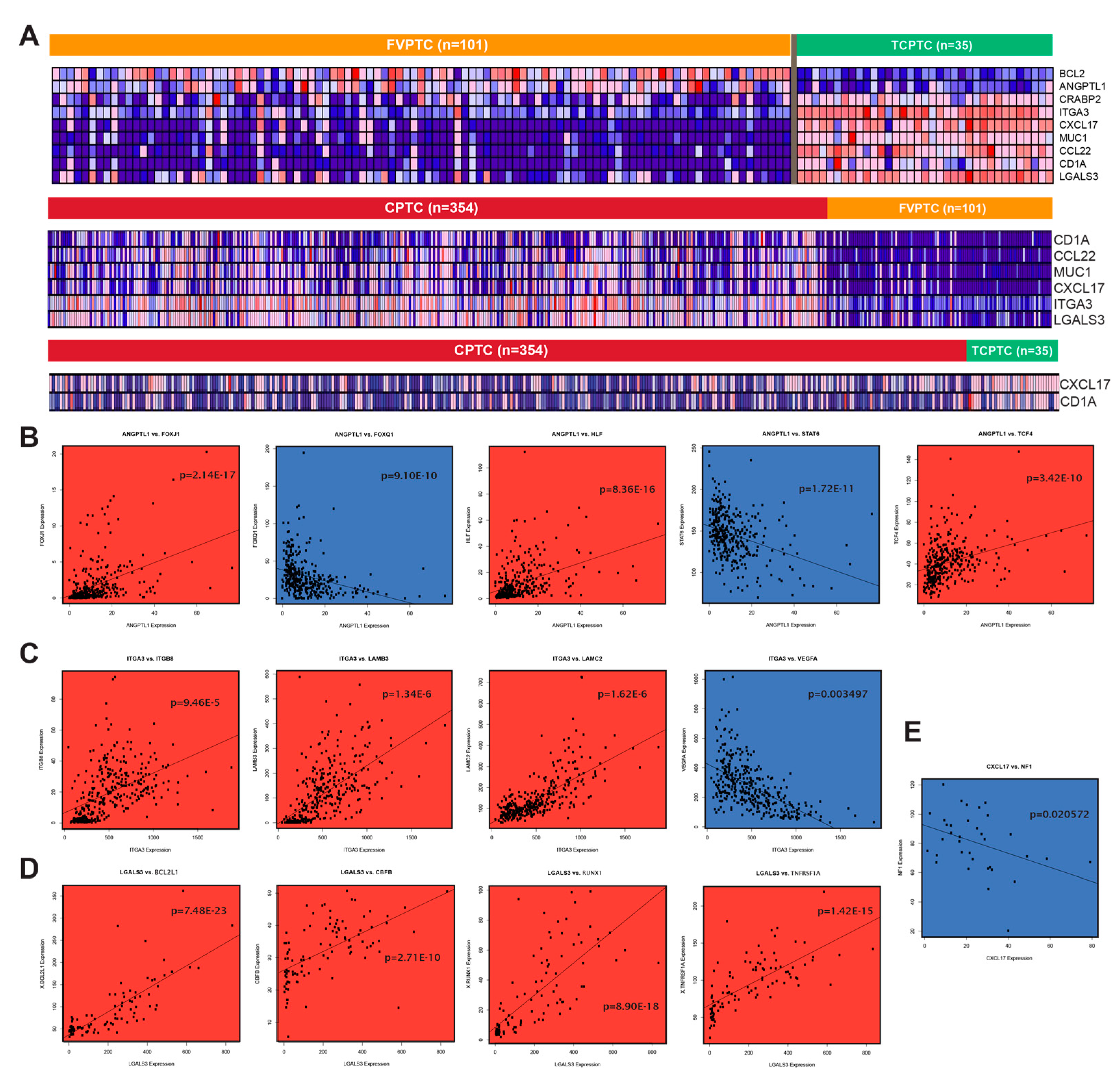
2.1. Similarities and differences between gene dysregulation landscapes of PTC subtypes
2.2. Filtering differentially expressed genes by MACIS score
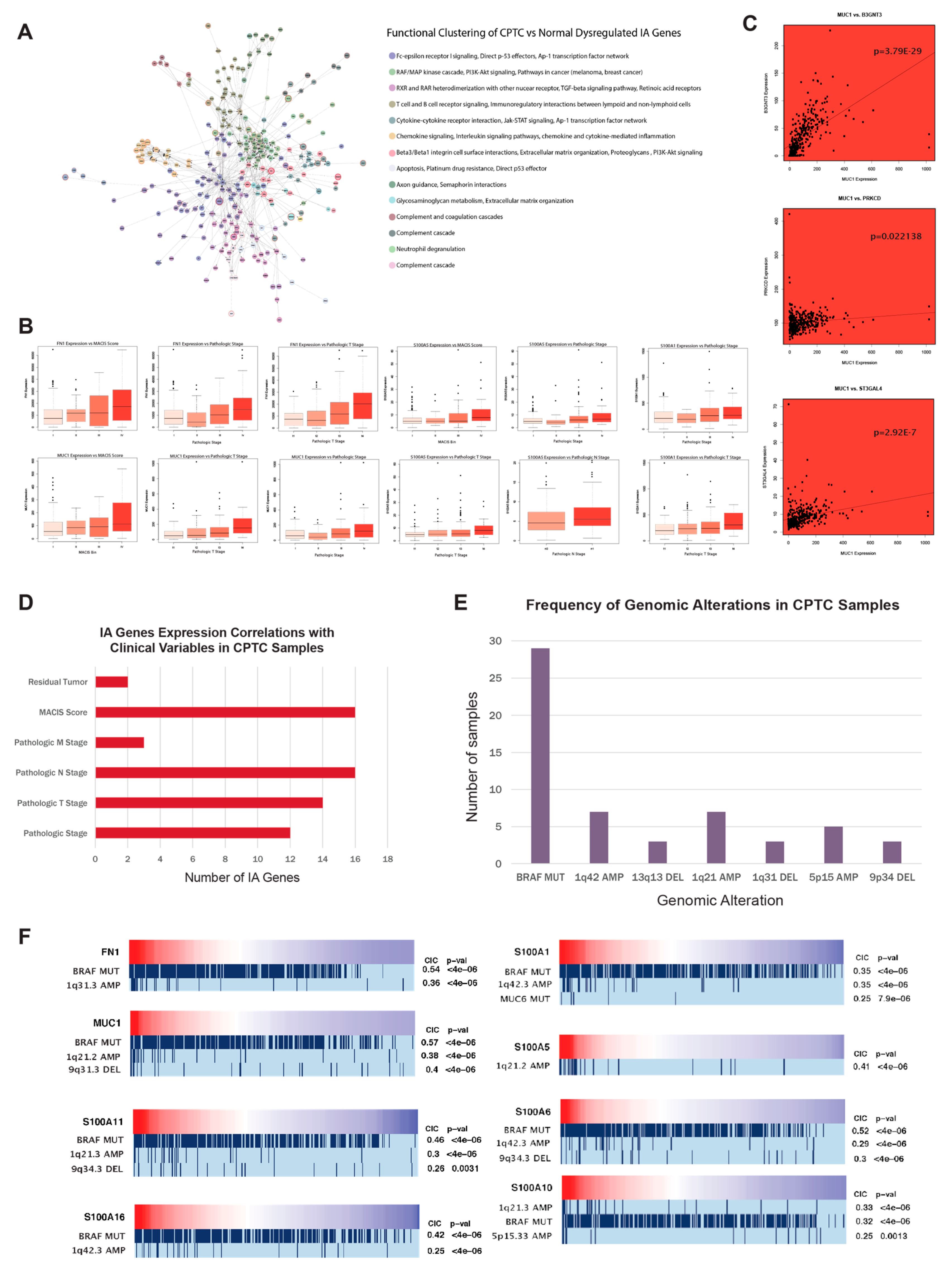
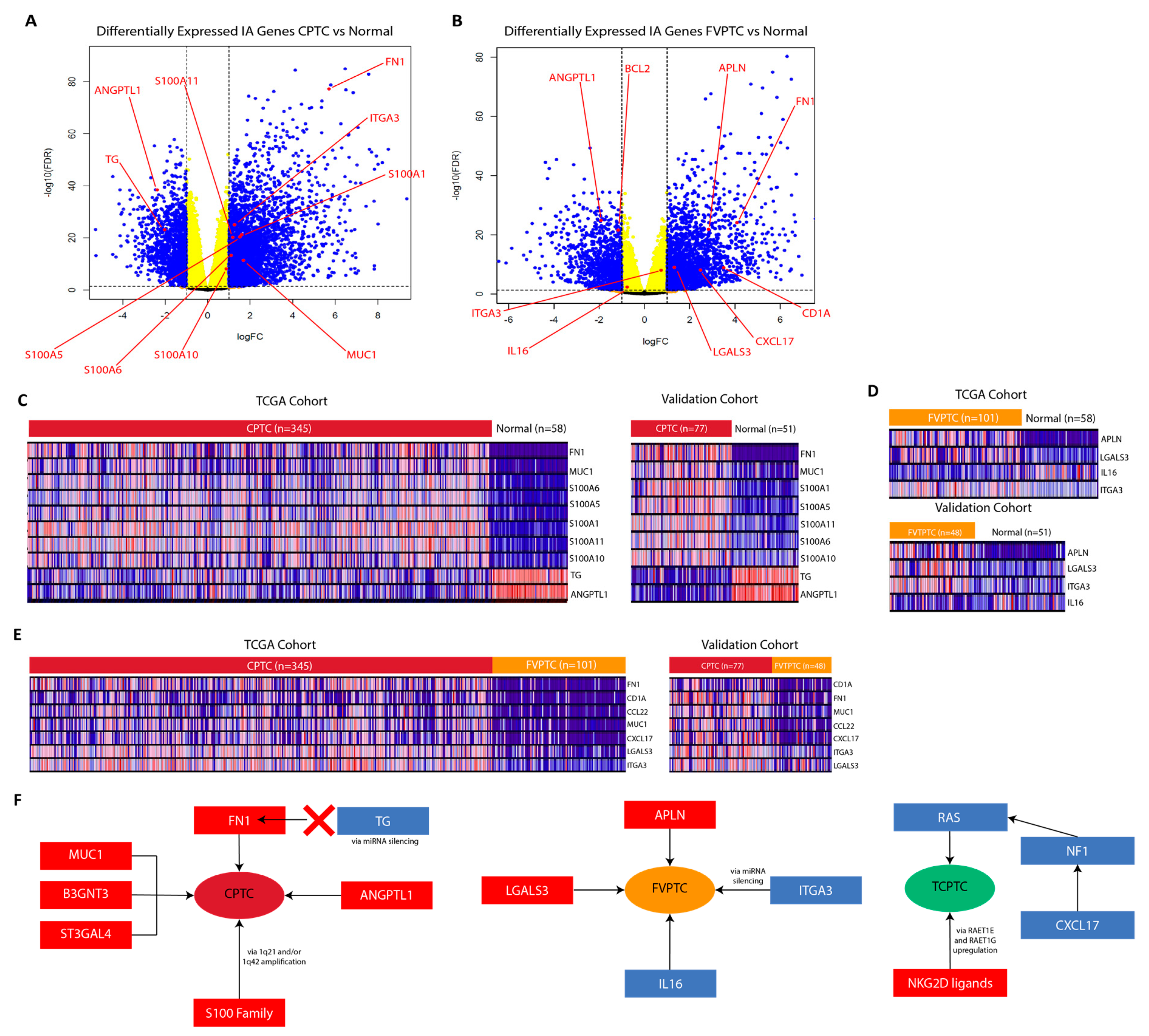
2.3. MUC1, FN1, S100 family genes, and CA/IA pathways define the CPTC dysregulation landscape
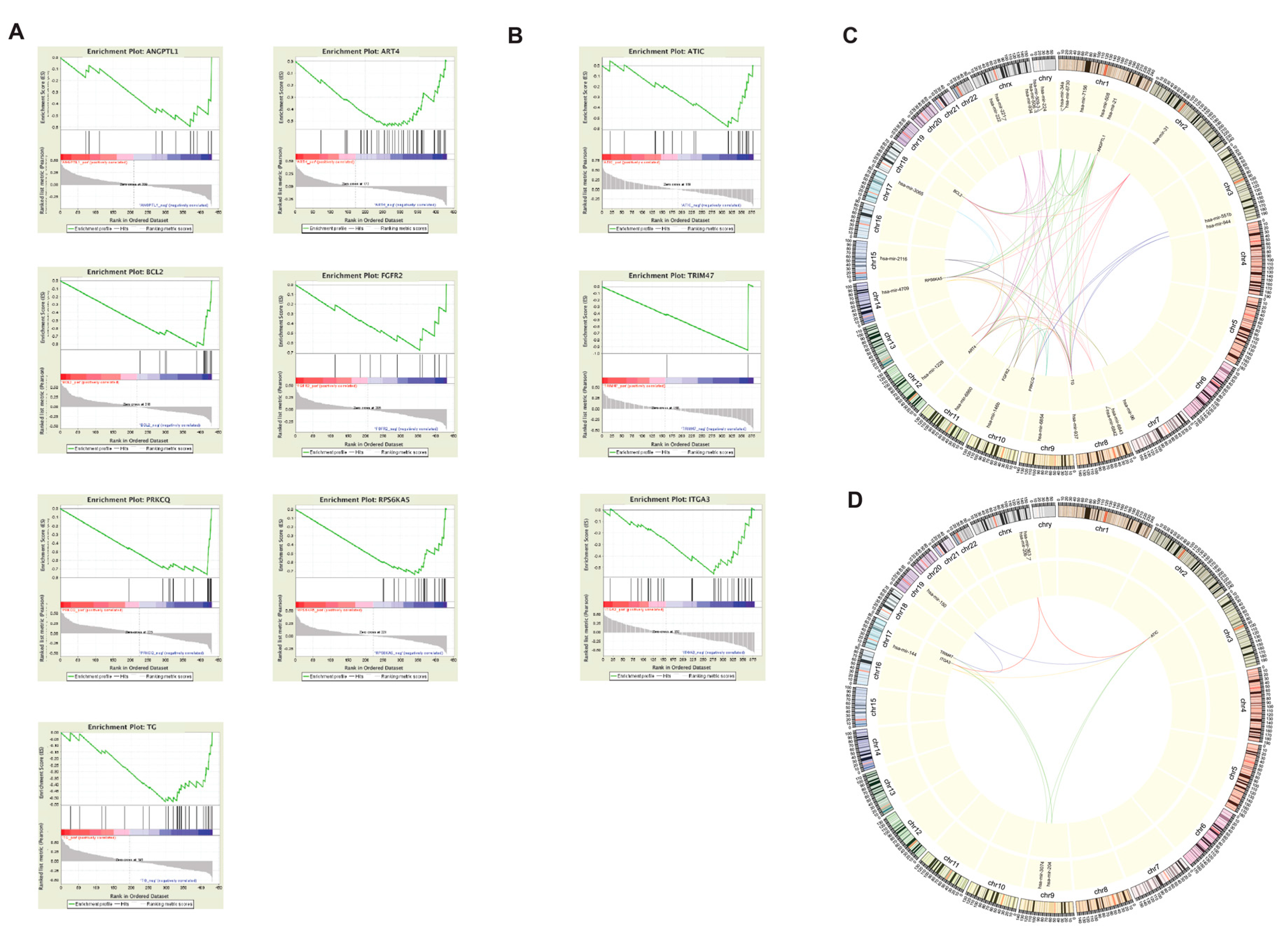
2.4. MUC1, FN1, and S100 family genes in CPTC correlated to alterations at tumor-associated loci
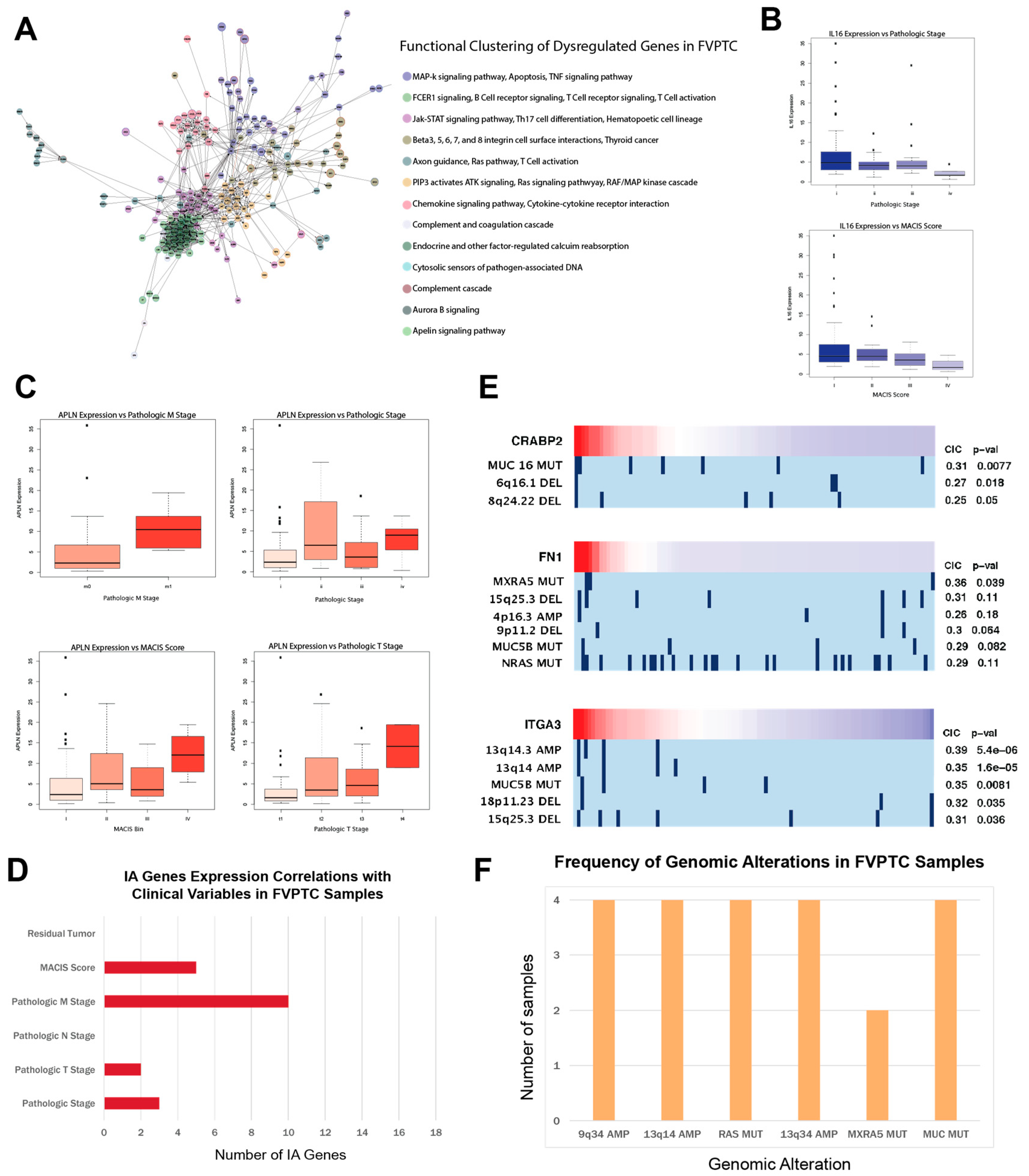
2.5. APLN, IL16, and CA/IA pathways define the FVPTC dysregulation landscape
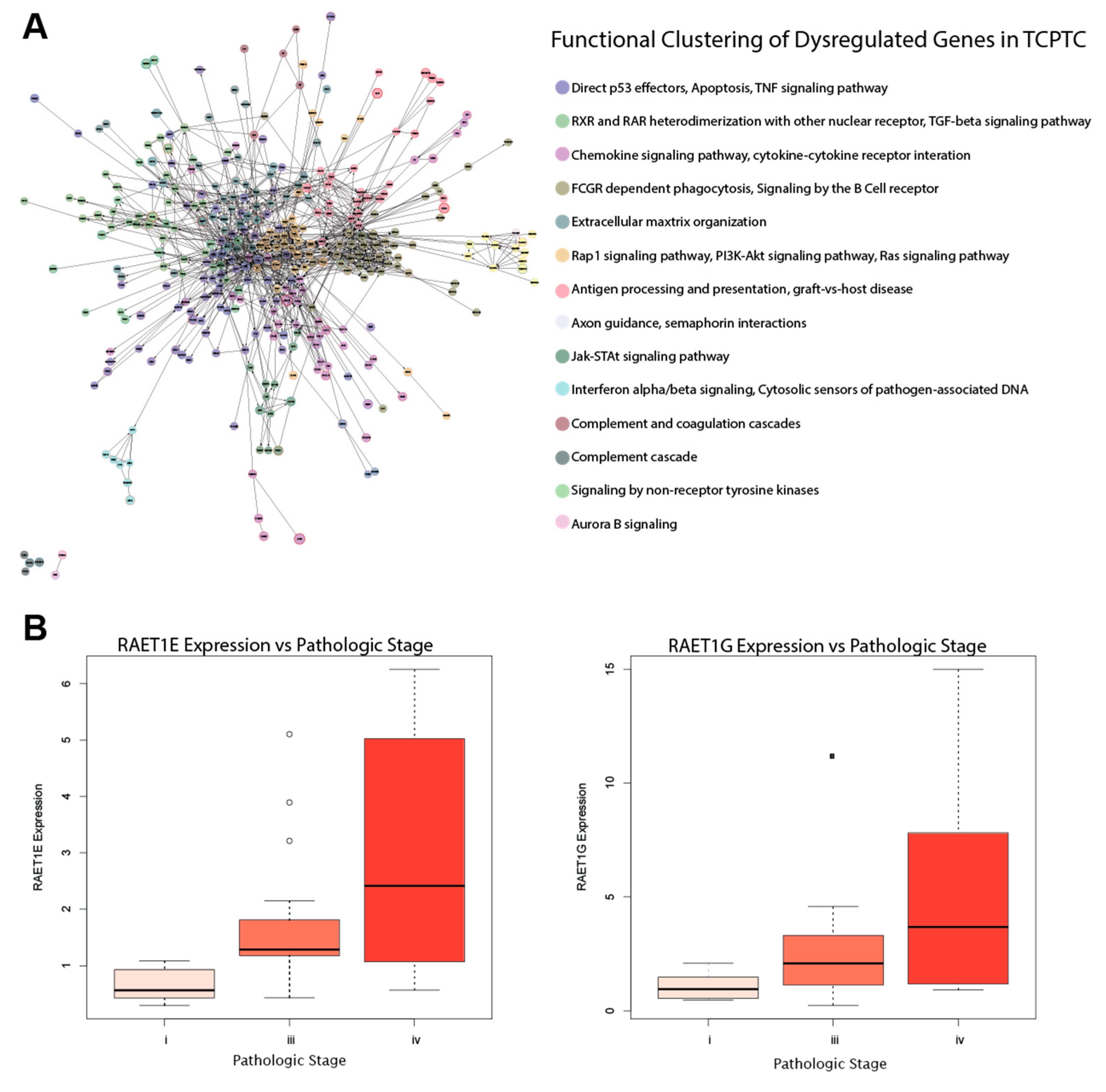
2.6. RAET-Family Genes and Antigen Presentation Pathways Define the TCPTC Dysregulation Landscape
2.7. Differentially expressed genes between PTC subtypes exhibit strong immune association
2.8. miRNA-mediated silencing of FN1 in CPTC and ITGA3 in FVPTC
2.9. Validation of RNA-sequencing data supports CPTC and FVPTC dysregulation patterns
3. Discussion
4. Materials and Methods
4.1. The Cancer Genome Atlas (TCGA) RNA-sequencing datasets and cohort designation
4.2. Differential expression analysis to identify dysregulated immune-associated genes
4.3. Functional Clustering Via ReactomeFIViz
4.4. Comparing gene expression to clinical variables
4.5. Correlating gene expression with genomic alterations
4.6. Association of gene expression with microRNA silencing
4.7. Determination of co-Expression between dysregulated genes and related genes
4.8. Validation of TCGA RNA-sequencing datasets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cramer, J.D.; Fu, P.; Harth, K.C.; Margevicius, S.; Wilhelm, S.M. Analysis of the rising incidence of thyroid cancer using the Surveillance, Epidemiology and End Results national cancer data registry. Surgery 2010, 148, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Liu, R.; Basolo, F.; Giannini, R.; Shen, X.; Teng, D.; Guan, H.; Shan, Z.; Teng, W.; Musholt, T.J.; et al. Differential Clinicopathological Risk and Prognosis of Major Papillary Thyroid Cancer Variants. J. Clin. Endocrinol. Metab. 2016, 101, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Vivero, M.; Kraft, S.; Barletta, J.A. Risk stratification of follicular variant of papillary thyroid carcinoma. Thyroid 2013, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Barletta, J.A. A user’s guide to non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP). Histopathology 2018, 72, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of Coexistent BRAF(V600E) and TERT Promoter Mutations on Poor Clinical Outcomes in Papillary Thyroid Cancer: A Meta-Analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef]
- Liu, D.; Hou, P.; Liu, Z.; Wu, G.; Xing, M. Genetic alterations in the phosphoinositide 3-kinase/Akt signaling pathway confer sensitivity of thyroid cancer cells to therapeutic targeting of Akt and mammalian target of rapamycin. Cancer Res. 2009, 69, 7311–7319. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. American Thyroid Association Guidelines Taskforce on Thyroid. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef]
- Chen, H.; Xu, C.; Jin, Q.; Liu, Z. S100 protein family in human cancer. Am. J. Cancer Res. 2014, 4, 89–115. [Google Scholar]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Soikkeli, J.; Podlasz, P.; Yin, M.; Nummela, P.; Jahkola, T.; Virolainen, S.; Krogerus, L.; Heikkila, P.; von Smitten, K.; Saksela, O.; et al. Metastatic outgrowth encompasses COL-I, FN1, and POSTN up-regulation and assembly to fibrillar networks regulating cell adhesion, migration, and growth. Am. J. Pathol. 2010, 177, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Pool, C.; Walter, V.; Bann, D.; Goldenberg, D.; Broach, J.; Hennessy, M.; Cottrill, E.; Washburn, E.; Williams, N.; Crist, H.; et al. Molecular characterization of tumors meeting diagnostic criteria for the non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP). Virchows Arch. 2019, 474, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A Cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar] [CrossRef]
- Renaud, F.; Gnemmi, V.; Devos, P.; Aubert, S.; Crepin, M.; Coppin, L.; Ramdane, N.; Bouchindhomme, B.; d’Herbomez, M.; Van Seuningen, I.; et al. MUC1 expression in papillary thyroid carcinoma is associated with BRAF mutation and lymph node metastasis; the latter is the most important risk factor of relapse. Thyroid 2014, 24, 1375–1384. [Google Scholar] [CrossRef]
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; De Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol. Cell Endocrinol. 2016, 431, 123–132. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Kim, J.W.; Botvinnik, O.B.; Abudayyeh, O.; Birger, C.; Rosenbluh, J.; Shrestha, Y.; Abazeed, M.E.; Hammerman, P.S.; DiCara, D.; Konieczkowski, D.J.; et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat. Biotechnol. 2016, 34, 539–546. [Google Scholar] [CrossRef]
- Patel, K.N.; Maghami, E.; Wreesmann, V.B.; Shaha, A.R.; Shah, J.P.; Ghossein, R.; Singh, B. MUC1 plays a role in tumor maintenance in aggressive thyroid carcinomas. Surgery 2005, 138, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Schaid, D.J.; Smith, J.R.; French, A.J.; Schroeder, J.J.; McDonnell, S.K.; Peterson, B.J.; Wang, Z.Y.; Carpten, J.D.; Roberts, S.G.; et al. Linkage analyses at the chromosome 1 loci 1q24-25 (HPC1), 1q42.2-43 (PCAP), and 1p36 (CAPB) in families with hereditary prostate cancer. Am. J. Hum. Genet. 2000, 66, 539–546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, J.; Xu, M.; Makowski, M.M.; Zhang, T.; Law, M.H.; Kovacs, M.A.; Granzhan, A.; Kim, W.J.; Parikh, H.; Gartside, M.; et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nat. Genet. 2017, 49, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; Hung, R.J.; Gaborieau, V.; Boffetta, P.; Chabrier, A.; Byrnes, G.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; et al. Lung cancer susceptibility locus at 5p15.33. Nat. Genet. 2008, 40, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Hornigold, N.; Devlin, J.; Davies, A.M.; Aveyard, J.S.; Habuchi, T.; Knowles, M.A. Mutation of the 9q34 gene TSC1 in sporadic bladder cancer. Oncogene 1999, 18, 2657–2661. [Google Scholar] [CrossRef] [PubMed]
- Mertens, D.; Wolf, S.; Tschuch, C.; Mund, C.; Kienle, D.; Ohl, S.; Schroeter, P.; Lyko, F.; Dohner, H.; Stilgenbauer, S.; et al. Allelic silencing at the tumor-suppressor locus 13q14.3 suggests an epigenetic tumor-suppressor mechanism. Proc. Natl. Acad. Sci. 2006, 103, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Schmidt, A.P.; Pashia, M.E.; Sunwoo, J.B.; Scholnick, S.B. Multiple regions of deletion on chromosome arm 13q in head-and-neck squamous-cell carcinoma. Int. J. Cancer 1999, 84, 453–457. [Google Scholar] [CrossRef]
- Ji, X.; Gui, J.; Han, Y.; Brennan, P.; Li, Y.; McKay, J.; Caporaso, N.E.; Bertazzi, P.A.; Landi, M.T.; Amos, C.I. The role of haplotype in 15q25.1 locus in lung cancer risk: results of scanning chromosome 15. Carcinogenesis 2015, 36, 1275–1283. [Google Scholar] [CrossRef][Green Version]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, X.X.; Xu, H.; Fang, H.Z.; Zhao, J.Q. Expression and prognostic significance of unique ULBPs in pancreatic cancer. Onco. Targets. Ther. 2016, 9, 5271–5279. [Google Scholar] [CrossRef]
- McGilvray, R.W.; Eagle, R.A.; Rolland, P.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int. J. Cancer 2010, 127, 1412–1420. [Google Scholar] [CrossRef]
- Stephen, J.K.; Chitale, D.; Narra, V.; Chen, K.M.; Sawhney, R.; Worsham, M.J. DNA methylation in thyroid tumorigenesis. Cancers 2011, 3, 1732–1743. [Google Scholar] [CrossRef]
- Koshizuka, K.; Hanazawa, T.; Kikkawa, N.; Arai, T.; Okato, A.; Kurozumi, A.; Kato, M.; Katada, K.; Okamoto, Y.; Seki, N. Regulation of ITGA3 by the anti-tumor miR-199 family inhibits cancer cell migration and invasion in head and neck cancer. Cancer Sci. 2017, 108, 1681–1692. [Google Scholar] [CrossRef]
- Yoo, S.K.; Lee, S.; Kim, S.J.; Jee, H.G.; Kim, B.A.; Cho, H.; Song, Y.S.; Cho, S.W.; Won, J.K.; Shin, J.Y.; et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS. Genet. 2016, 12, e1006239. [Google Scholar] [CrossRef]
- National Cancer Institute. Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov (accessed on 8 July 2019).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Dunn, P.; Thomas, C.G.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data 2018, 5, 180015. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Olsen, L.R.; Tongchusak, S.; Lin, H.; Reinherz, E.L.; Brusic, V.; Zhang, G.L. TANTIGEN: A comprehensive database of tumor T cell antigens. Cancer Immunol. Immunother. 2017, 66, 731–735. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, F.; Wei, S.; Xu, G. Identification of Key Genes Affecting Results of Hyperthermia in Osteosarcoma Based on Integrative ChIP-Seq/TargetScan Analysis. Med. Sci. Monit. 2017, 23, 2042–2048. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakladar, J.; Li, W.T.; Bouvet, M.; Chang, E.Y.; Wang-Rodriguez, J.; Ongkeko, W.M. Papillary Thyroid Carcinoma Variants are Characterized by Co-dysregulation of Immune and Cancer Associated Genes. Cancers 2019, 11, 1179. https://doi.org/10.3390/cancers11081179
Chakladar J, Li WT, Bouvet M, Chang EY, Wang-Rodriguez J, Ongkeko WM. Papillary Thyroid Carcinoma Variants are Characterized by Co-dysregulation of Immune and Cancer Associated Genes. Cancers. 2019; 11(8):1179. https://doi.org/10.3390/cancers11081179
Chicago/Turabian StyleChakladar, Jaideep, Wei Tse Li, Michael Bouvet, Eric Y. Chang, Jessica Wang-Rodriguez, and Weg M. Ongkeko. 2019. "Papillary Thyroid Carcinoma Variants are Characterized by Co-dysregulation of Immune and Cancer Associated Genes" Cancers 11, no. 8: 1179. https://doi.org/10.3390/cancers11081179
APA StyleChakladar, J., Li, W. T., Bouvet, M., Chang, E. Y., Wang-Rodriguez, J., & Ongkeko, W. M. (2019). Papillary Thyroid Carcinoma Variants are Characterized by Co-dysregulation of Immune and Cancer Associated Genes. Cancers, 11(8), 1179. https://doi.org/10.3390/cancers11081179