Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles




Abstract
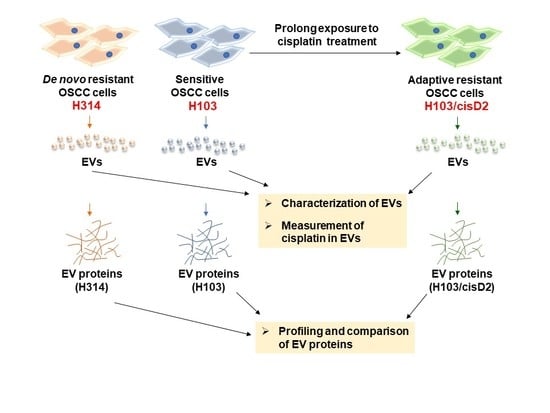
1. Introduction
2. Results
2.1. Screening and Establishment of Resistant Cell Lines
2.2. Characterization of OSCC Cell-Derived EVs
2.3. Protein Profiling of OSCC Cell-Derived EVs
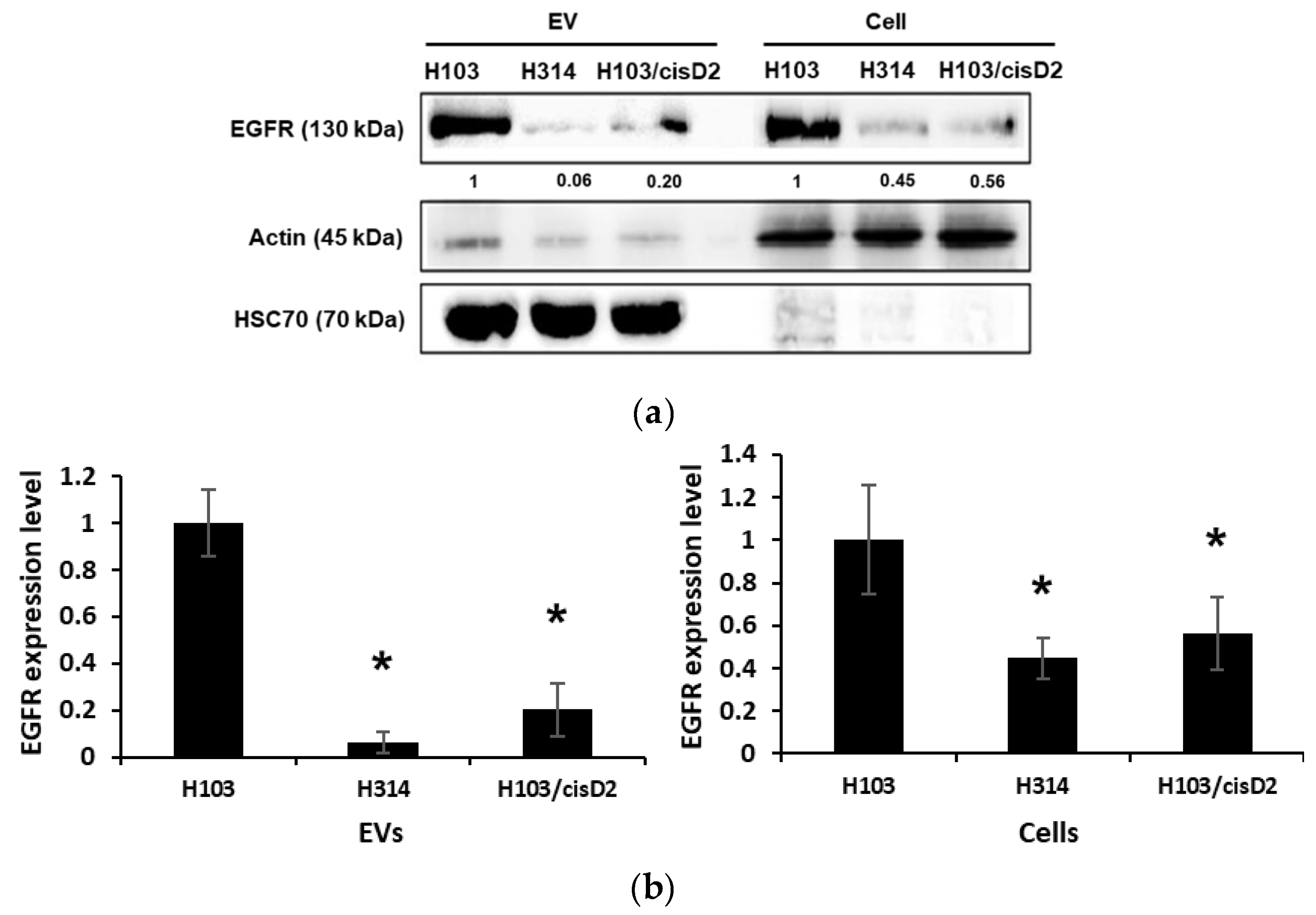
2.4. Regulation of EGFR in OSCC Cell-Derived EVs
2.5. Regulation of Drug Resistance-Associated Proteins in OSCC Cell-Derived EVs
2.6. Effect of EV Inhibition on Drug Response of OSCC Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Maintenance
4.3. Cell Viability Assay
4.4. Establishment of Cisplatin-Resistant Cell Line
4.5. EV Isolation
4.6. Transmission Electron Microscopy (TEM)
4.7. Nanoparticle Tracking Analysis (NTA)
4.8. Inductively-Coupled Plasma Mass Spectrometry (ICP-MS)
4.9. Protein Lysis and Digestion
4.10. Liquid Chromatography-Mass Spectrometry/Mass Spectrometry (LCMS/MS)
4.11. Protein Identification
4.12. Protein Network Analysis
4.13. Western Blot Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA Cancer J. Clin. 2009, 52, 195–215. [Google Scholar] [CrossRef]
- Kernohan, M.D.; Clark, J.R.; Gao, K.; Ebrahimi, A.; Milross, C.G. Predicting the prognosis of oral squamous cell carcinoma after first recurrence. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Dalton, W.S. De novo and acquired resistance to antitumor alkylating agents. In Cancer Drug Resistance; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 377–389. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, G.; Sun, D.; Lei, M.; Li, Y.; Zhou, C.; Li, X.; Xue, W.; Wang, H.; Liu, C.; et al. Exosomes containing miR-21 transfer the characteristic of cisplatin resistance by targeting PTEN and PDCD4 in oral squamous cell carcinoma. Acta Biochim. Biophys. Sin. 2017, 49, 808–816. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- Pérez-Sayáns, M.; Somoza-Martín, J.M.; Barros-Angueira, F.; Diz, P.G.; Rey, J.M.G.; García-García, A. Multidrug Resistance in Oral Squamous Cell Carcinoma: The Role of Vacuolar ATPases. Cancer Lett. 2010, 295, 135–143. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental PH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Rodríguez Zorrilla, S.; Pérez-Sayans, M.; Fais, S.; Logozzi, M.; Gallas Torreira, M.; García García, A. A Pilot Clinical Study on the Prognostic Relevance of Plasmatic Exosomes Levels in Oral Squamous Cell Carcinoma Patients. Cancers 2019, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Prime, S.S.; Nixon, S.V.; Crane, I.J.; Stone, A.; Matthews, J.B.; Maitland, N.J.; Remnant, L.; Powell, S.K.; Game, S.M.; Scully, C. The behaviour of human oral squamous cell carcinoma in cell culture. J. Pathol. 1990, 160, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Edington, K.G.; Loughran, O.P.; Berry, I.J.; Parkinson, E.K. Cellular immortality: A late event in the progression of human squamous cell carcinoma of the head and neck associated with p53 alteration and a high frequency of allele loss. Mol. Carcinog. 1995, 13, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef]
- Rana, S.; Zöller, M. The functional importance of tetraspanins in exosomes. In Emerging Concepts of Tumor Exosomes-Mediated Cell-Cell Communication; Zhang, H.G., Ed.; Springer: New York, NY, USA, 2013; pp. 69–106. [Google Scholar] [CrossRef]
- Chiasson-MacKenzie, C.; McClatchey, A.I. EGFR-induced cytoskeletal changes drive complex cell behaviors: The tip of the iceberg. Sci. Signal. 2018, 11, eaas9473. [Google Scholar] [CrossRef]
- Kimura, I.; Kitahara, H.; Ooi, K.; Kato, K.; Noguchi, N.; Yoshizawa, K.; Nakamura, H.; Kawashiri, S. Loss of epidermal growth factor receptor expression in oral squamous cell carcinoma is associated with invasiveness and epithelial-mesenchymal transition. Oncol. Lett. 2016, 11, 201–207. [Google Scholar] [CrossRef]
- Lee, J.E.; Moon, P.G.; Cho, Y.E.; Kim, Y.B.; Kim, I.S.; Park, H.; Baek, M.C. Identification of EDIL3 on extracellular vesicles involved in breast cancer cell invasion. J. Proteom. 2016, 131, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Beckham, C.J.; Olsen, J.; Yin, P.N.; Wu, C.H.; Ting, H.J.; Hagen, F.K.; Scosyrev, E.; Messing, E.M.; Lee, Y.F. Bladder cancer exosomes contain EDIL-3/Del1 and facilitate cancer progression. J. Urol. 2014, 192, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; McCarty, N. Tampering with cancer chemoresistance by targeting the TGM2-IL6-autophagy regulatory network. Autophagy 2017, 13, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Kim, W.J.; Demircan, B.; Dyer, L.M.; Bray, K.J.; Skehan, R.R.; Massoll, N.A.; Brown, K.D. The transglutaminase 2 gene (TGM2), a potential molecular marker for chemotherapeutic drug sensitivity, is epigenetically silenced in breast cancer. Carcinogenesis 2008, 29, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 2016, 1863, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Kawazoe, Y.; Ikeno, M.; Saitoh, M.; Nakano, Y.; Nishi, Y.; Fukushima, S.; Takeuchi, Y. Role of Na+, K+-ATPase alpha1 subunit in the intracellular accumulation of cisplatin. Cancer Chemother. Pharmacol. 2006, 57, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.X.; Liu, X.M.; Lv, M.M.; Chen, L.; Zhao, J.H.; Zhong, S.L.; Ji, M.H.; Hu, Q.; Luo, Z.; Wu, J.Z.; et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef]
- Guan, X.W.; Zhao, F.; Wang, J.Y.; Wang, H.Y.; Ge, S.H.; Wang, X.; Zhang, L.; Liu, R.; Ba, Y.; Li, H.L.; et al. Tumor microenvironment interruption: A novel anti-cancer mechanism of proton-pump inhibitor in gastric cancer by suppressing the release of microRNA-carrying exosomes. Am. J. Cancer Res. 2017, 7, 1913–1925. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EVs Derived from Cell Lines | Size of EVs (nm) | |||||
|---|---|---|---|---|---|---|
| Mode | Mean | a SD | b D10 | c D50 | d D90 | |
| H103 | 103.6 ± 5.6 | 111.6 ± 17.9 | 46.1 ± 5.6 | 65.5 ± 5.7 | 97.1 ± 7.9 | 158.5 ± 22.2 |
| H314 | 100.2 ± 12.5 | 130.0 ± 9.7 | 46.7 ± 11.4 | 78.4 ± 4.5 | 107.8 ± 4.3 | 179.7 ± 16.9 |
| H103/cisD2 | 82.7 ± 22.5 | 99.8 ± 16.3 | 37.1 ± 1.0 | 55.0 ± 12.6 | 81.76 ± 19.8 | 132.4 ± 14.4 |
| Cell Line | Mean Growth Rate (h−1) | Doubling Time (h) |
|---|---|---|
| H103 | 0.0343 | 20.2 ± 3.9 a |
| H314 | 0.0204 | 33.9 ± 1.4 b |
| H103/cisD2 | 0.0259 | 26.9 ± 1.8 c |
| Proteins | 1 Fold Change in EV | Function | |
|---|---|---|---|
| H314 | H103/cisD2 | ||
| PACSIN3 | −5.57688 | −2.74937 | Plays a role in endocytosis and regulates internalization of plasma membrane proteins |
| CTNNB1 | −6.31522 | −2.66626 | Part of a complex of proteins that constitute adherens junctions |
| DLG1 | −4.16456 | −4.03571 | May have a role in septate junction formation, signal transduction, cell proliferation, synaptogenesis and lymphocyte activation. |
| ATP1A1 | −1.61594 | −1.30331 | Maintaining the electrochemical gradients of Na and K ions across the plasma membrane |
| ATP1B3 | −1.87647 | −1.651 | Maintaining the electrochemical gradients of Na and K ions across the plasma membrane |
| CAB39 | −2.64897 | −1.95177 | Component of a complex that binds and activates STK11/LKB1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoo, X.-H.; Paterson, I.C.; Goh, B.-H.; Lee, W.-L. Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles. Cancers 2019, 11, 1166. https://doi.org/10.3390/cancers11081166
Khoo X-H, Paterson IC, Goh B-H, Lee W-L. Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles. Cancers. 2019; 11(8):1166. https://doi.org/10.3390/cancers11081166
Chicago/Turabian StyleKhoo, Xin-Hui, Ian C. Paterson, Bey-Hing Goh, and Wai-Leng Lee. 2019. "Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles" Cancers 11, no. 8: 1166. https://doi.org/10.3390/cancers11081166
APA StyleKhoo, X.-H., Paterson, I. C., Goh, B.-H., & Lee, W.-L. (2019). Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles. Cancers, 11(8), 1166. https://doi.org/10.3390/cancers11081166