CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs


Abstract
1. Introduction
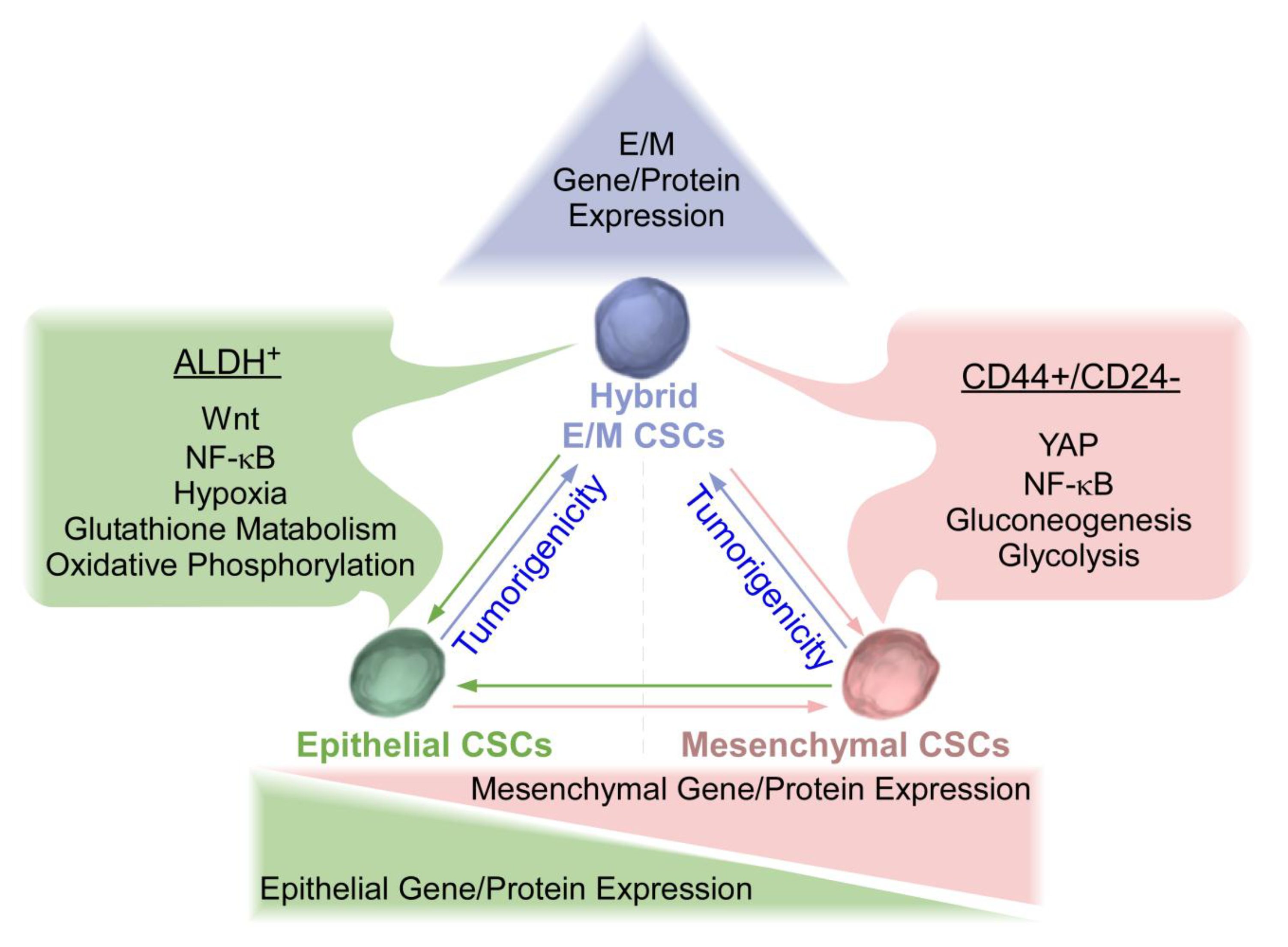
2. Overview of Epithelial and Mesenchymal CSCs in Breast Cancer
3. Wnt/β-Catenin Signaling in Breast Cancer and TNBC and Its Association with Epithelial CSCs
4. YAP Signaling in Breast Cancer and TNBC and Its Association with M CSCs
5. NF-κB, Cytokines and the Tumor Microenvironment in M and E CSCs
6. Hypoxia Signaling in Breast Cancer and TNBC E CSCs
7. Future Directions
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH | aldehyde dehydrogenase |
| CD44 | cluster of differentiation 44 (hyaluronic acid receptor) |
| CD24 | cluster of differentiation 24 |
| CSC | cancer stem cell |
| PDX | patient derived xenograft |
| TNBC | triple negative breast cancer |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Shi, L.H.; Wang, X.J.; Wang, S.X.; Wan, X.Q.; Liu, S.R.; Wang, Y.F.; Lu, Z.; Wang, L.H.; Ding, Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018, 53, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Jin, Y.; Zhang, D.; Lin, D. 5-Fluorouracil may enrich cancer stem cells in canine mammary tumor cells in vitro. Oncol. Lett. 2018, 15, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yuan, J.; Yuan, S.; Yang, Y.; Feng, X.; Niu, Y. Survival estimates based on molecular subtype and age in patients with early node-negative breast cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 5357–5367. [Google Scholar]
- Matsuda, S.; Yan, T.; Mizutani, A.; Sota, T.; Hiramoto, Y.; Prieto-Vila, M.; Chen, L.; Satoh, A.; Kudoh, T.; Kasai, T. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int. J. Cancer 2014, 135, 27–36. [Google Scholar] [CrossRef]
- ElShamy, W.M.; Duhé, R.J. Overview: Cellular plasticity, cancer stem cells and metastasis. Cancer Lett. 2013, 341, 2–8. [Google Scholar] [CrossRef]
- Liu, P.; Kumar, I.; Brown, S.; Kannappan, V.; Tawari, P.; Tang, J.; Jiang, W.; Armesilla, A.; Darling, J.; Wang, W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br. J. Cancer 2013, 109, 1876. [Google Scholar] [CrossRef]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- El-Badawy, A.; Ghoneim, N.I.; Nasr, M.A.; Elkhenany, H.; Ahmed, T.A.; Ahmed, S.M.; El-Badri, N. Telomerase reverse transcriptase coordinates with the epithelial-to-mesenchymal transition through a feedback loop to define properties of breast cancer stem cells. Biol. Open 2018, 7, bio.034181. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching moving targets: Cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 2017, 16, 43. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as Cancer Stem Cell Markers: An Enduring Ambiguity. Clin. Dev. Immunol. 2012, 2012, 708036. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin. Cancer Res. 2010, 16, 876–887. [Google Scholar] [CrossRef]
- Kastan, M.B.; Schlaffer, E.; Russo, J.E.; Colvin, O.M.; Civin, C.I.; Hilton, J. Direct demonstration of elevated aldehyde dehydrogenase in human hematopoietic progenitor cells. Blood 1990, 75, 1947–1950. [Google Scholar]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C.J. Crio: On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Khaled, N.; Bidet, Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers 2019, 11, 559. [Google Scholar] [CrossRef]
- Sulaiman, A.; Yao, Z.; Wang, L. Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression. J. Biomed. Res. 2018, 32, 81. [Google Scholar]
- Bastid, J. EMT in carcinoma progression and dissemination: Facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012, 31, 277–283. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1. [Google Scholar] [CrossRef]
- Xue, C.; Plieth, D.; Venkov, C.; Xu, C.; Neilson, E.G. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003, 63, 3386–3394. [Google Scholar]
- Fischer, K.R.; Altorki, N.K.; Mittal, V.; Gao, D. Fischer et al. reply. Nature 2017, 547, E5. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; d’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nature 2018, 20, 1349. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209. [Google Scholar] [CrossRef]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/β-catenin Pathway Activation Is Enriched in Basal-Like Breast Cancers and Predicts Poor Outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef]
- Kim, T.; Yang, S.J.; Hwang, D.; Song, J.; Kim, M.; Kim, S.K.; Kang, K.; Ahn, J.; Lee, D.; Kim, M.Y.; et al. A basal-like breast cancer-specific role for SRF–IL6 in YAP-induced cancer stemness. Nat. Commun. 2015, 6, 10186. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Nam, J.S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers 2019, 11, 965. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef]
- Bhavanasi, D.; Klein, P.S. Wnt Signaling in Normal and Malignant Stem Cells. Curr. Stem Cell Rep. 2016, 2, 379–387. [Google Scholar] [CrossRef]
- Yu, Q.C.; Verheyen, E.M.; Zeng, Y.A. Mammary Development and Breast Cancer: A Wnt Perspective. Cancers 2016, 8, 65. [Google Scholar] [CrossRef]
- Xu, W.H.; Liu, Z.B.; Yang, C.; Qin, W.; Shao, Z.M. Expression of Dickkopf-1 and Beta-catenin Related to the Prognosis of Breast Cancer Patients with Triple Negative Phenotype. PLoS ONE 2012, 7, e37624. [Google Scholar] [CrossRef]
- Forget, M.A.; Turcotte, S.; Beauseigle, D.; Godin-Ethier, J.; Pelletier, S.; Martin, J.; Tanguay, S.; Lapointe, R. The Wnt pathway regulator DKK1 is preferentially expressed in hormone-resistant breast tumours and in some common cancer types. Br. J. Cancer 2007, 96, 646–653. [Google Scholar] [CrossRef]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef]
- Tzeng, H.E.; Yang, L.; Chen, K.; Wang, Y.; Liu, Y.R.; Pan, S.L.; Gaur, S.; Hu, S.; Yen, Y. The pan-PI3K inhibitor GDC-0941 activates canonical WNT signaling to confer resistance in TNBC cells: Resistance reversal with WNT inhibitor. Oncotarget 2015, 6, 11061–11073. [Google Scholar] [CrossRef]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef]
- Howe, L.R.; Brown, A.M. Wnt signaling and breast cancer. Cancer Biol. Ther. 2004, 3, 36–41. [Google Scholar] [CrossRef]
- Wend, P.; Runke, S.; Wend, K.; Anchondo, B.; Yesayan, M.; Jardon, M.; Hardie, N.; Loddenkemper, C.; Ulasov, I.; Lesniak, M.S.; et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013, 5, 264–279. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef]
- Jung, H.Y.; Jun, S.; Lee, M.; Kim, H.C.; Wang, X.; Ji, H.; McCrea, P.D.; Park, J.I. PAF and EZH2 Induce Wnt/β-Catenin Signaling Hyperactivation. Mol. Cell 2013, 52, 193–205. [Google Scholar] [CrossRef]
- El Ayachi, I.; Fatima, I.; Wend, P.; Alva-Ornelas, J.A.; Runke, S.; Kuenzinger, W.L.; Silva, J.; Silva, W.; Gray, J.K.; Lehr, S. The WNT10B network is associated with survival and metastases in chemoresistant triple-negative breast cancer. Cancer Res. 2019, 79, 982–993. [Google Scholar] [CrossRef]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34, e15721. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z.; et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef]
- Domenici, G.; Aurrekoetxea-Rodríguez, I.; Simões, B.M.; Rábano, M.; Lee, S.Y.; San Millán, J.; Comaills, V.; Oliemuller, E.; López-Ruiz, J.A.; Zabalza, I. A Sox2–Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151. [Google Scholar] [CrossRef]
- Wang, H.; He, L.; Ma, F.; Regan, M.M.; Balk, S.P.; Richardson, A.L.; Yuan, X. SOX9 Regulates Low Density Lipoprotein Receptor-related Protein 6 (LRP6) and T-cell Factor 4 (TCF4) Expression and Wnt/β-catenin Activation in Breast Cancer. J. Biol. Chem. 2013, 288, 6478–6487. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J.J.C. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Ring, A.; Nguyen, C.; Smbatyan, G.; Tripathy, D.; Yu, M.; Press, M.; Kahn, M.; Lang, J.E. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers 2018, 10, 525. [Google Scholar] [CrossRef]
- Diamond, J.R.; Andreopoulou, E.; Favret, A.M.; Nanda, R.; Peterson, C.; Benaim, E. 363TiPPhase Ib/IIa study of RX-5902, a novel orally bioavailable inhibitor of phosphorylated P68, which prevents nuclear β-catenin translocation in patients with triple negative breast cancer. Ann. Oncol. 2018, 29, 272–351. [Google Scholar] [CrossRef]
- Tentler, J.; Frank, J.; Kim, D.; George, C.; Lee, Y.; Ely, B.; Tan, A.; Kim, J.; Pitts, T.; Capasso, A.; et al. Abstract P5-21-16: Preclinical studies of RX-5902, a beta-catenin modulator in triple negative breast cancer. Cancer Res. 2018, 78, 5–21. [Google Scholar]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef]
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 93. [Google Scholar] [CrossRef]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.-W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef]
- Horst, D.; Chen, J.; Morikawa, T.; Ogino, S.; Kirchner, T.; Shivdasani, R.A. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer Res. 2012, 72, 1547–1556. [Google Scholar] [CrossRef]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Liang, X.; Sun, S.; Jiang, Y.; Xia, B.; Niu, M.; Li, D.; Zhang, J.; Wang, S.; et al. Androgen Receptor Expression and Bicalutamide Antagonize Androgen Receptor Inhibit β-catenin Transcription Complex in Estrogen Receptor-Negative Breast Cancer. Cell. Physiol. Biochem. 2017, 43, 2212–2225. [Google Scholar] [CrossRef]
- Clinical Trial Search. Available online: https://clinicaltrials.gov/ct2/results?cond=Triple+Negative+ Breast+Cancer&Search=Apply&recrs=d&age_v=&gndr=Female&type=Intr&rslt=&phase=0&phase=1&phase=2&phase=3 (accessed on 30 March 2019).
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Pan, D. Hippo signaling in organ size control. Genes Dev. 2007, 21, 886–897. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Vlug, E.J.; Van De Ven, R.A.H.; Vermeulen, J.F.; Bult, P.; Van Diest, P.J.; Derksen, P.W.B. Nuclear localization of the transcriptional coactivator YAP is associated with invasive lobular breast cancer. Cell. Oncol. 2013, 36, 375–384. [Google Scholar] [CrossRef]
- Yang, X.; Li, D.M.; Chen, W.; Xu, T. Human homologue of Drosophila lats, LATS1, negatively regulate growth by inducing G2/M arrest or apoptosis. Oncogene 2001, 20, 6516–6523. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, W.H.; Koo, J.S. Expression of Yes-associated protein (YAP) in metastatic breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11248–11257. [Google Scholar]
- Zhao, Y.; Khanal, P.; Savage, P.; She, Y.-M.; Cyr, T.D.; Yang, X. YAP-Induced Resistance of Cancer Cells to Antitubulin Drugs Is Modulated by a Hippo-Independent Pathway. Cancer Res. 2014, 74, 4493–4503. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; El-Sahli, S.; Li, L.; Chambers, J.; Phan, A.; Cote, M.; Cron, G.O.; Alain, T.; Le, Y.; et al. Co-Targeting Bulk Tumor and CSCs in Clinically Translatable TNBC Patient-Derived Xenografts via Combination Nanotherapy. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Zhang, R.; Xiao, W.; Niu, X.; Ye, X.; Li, Z.; Guo, Y.; Tan, J.; Li, Y. The EZH2- H3K27me3-DNMT1 complex orchestrates epigenetic silencing of the wwc1 gene, a Hippo/YAP pathway upstream effector, in breast cancer epithelial cells. Cell. Signal. 2018, 51, 243–256. [Google Scholar] [CrossRef]
- Campbell, M.J.; Esserman, L.J.; Zhou, Y.; Shoemaker, M.; Lobo, M.; Borman, E.; Baehner, F.; Kumar, A.S.; Adduci, K.; Marx, C. Breast cancer growth prevention by statins. Cancer Res. 2006, 66, 8707–8714. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Chan, S.W.; Lim, C.J.; Guo, K.; Ng, C.P.; Lee, I.; Hunziker, W.; Zeng, Q.; Hong, W. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008, 68, 2592–2598. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J. Unbalanced YAP–SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene 2018, 38, 2042–2055. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Ghia, E.M.; Huang, J.; Wu, L.; Zhang, J.; Lam, S.; Lei, Y.; He, J.; Cui, B. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 1370–1377. [Google Scholar] [CrossRef]
- Samanta, S.; Guru, S.; Elaimy, A.L.; Amante, J.J.; Ou, J.; Yu, J.; Zhu, L.J.; Mercurio, A.M. IMP3 Stabilization of WNT5B mRNA Facilitates TAZ Activation in Breast Cancer. Cell Rep. 2018, 23, 2559–2567. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nature 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Thongon, N.; Castiglioni, I.; Zucal, C.; Latorre, E.; D’Agostino, V.; Bauer, I.; Pancher, M.; Ballestrero, A.; Feldmann, G.; Nencioni, A.; et al. The GSK3β inhibitor BIS I reverts YAP-dependent EMT signature in PDAC cell lines by decreasing SMADs expression level. Oncotarget 2016, 7, 26551–26566. [Google Scholar] [CrossRef]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef]
- Kim, H.B.; Kim, M.; Park, Y.S.; Park, I.; Kim, T.; Yang, S.Y.; Cho, C.J.; Hwang, D.; Jung, J.H.; Markowitz, S.D. Prostaglandin E2 activates YAP and a positive-signaling loop to promote colon regeneration after colitis but also carcinogenesis in mice. Gastroenterology 2017, 152, 616–630. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, D.; Li, H.; Wang, L.; Tian, G.; Dong, Y. YAP overexpression promotes the epithelial-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol. Med. Rep. 2016, 13, 237–242. [Google Scholar] [CrossRef]
- Liu, F.; Wang, G.; Wang, X.; Che, Z.; Dong, W.; Guo, X.; Wang, Z.; Chen, P.; Hou, D.; Zhang, Q.; et al. Targeting high Aurora kinases expression as an innovative therapy for hepatocellular carcinoma. Oncotarget 2017, 8, 27953–27965. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Serasanambati, M.; Chilakapati, S.R. Function of nuclear factor kappa B (NF-κB) in human diseases-a review. S. Indian J. Biol. Sci. 2016, 2, 368–387. [Google Scholar]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Davis, J.N.; Kucuk, O.; Sarkar, F.H. Genistein inhibits NF-κB activation in prostate cancer cells. Nutr. Cancer 1999, 35, 167–174. [Google Scholar] [CrossRef]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther. 2014, 7, 1015. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; Hollander, P.D.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the pro-inflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Aryappalli, P.; Al-Qubaisi, S.S.; Attoub, S.; George, J.A.; Arafat, K.; Ramadi, K.B.; Mohamed, Y.A.; Al-Dhaheri, M.M.; Al-Sbiei, A.; Fernandez-Cabezudo, M.J.; et al. The IL-6/sTaT3 Signaling Pathway Is An Early Target of Manuka Honey-Induced Suppression of Human Breast Cancer Cells. Front. Oncol. 2017, 7, 167. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin. Transl. Med. 2018, 7, 27. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J.J.C. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef]
- Gallo, M.; Frezzetti, D.; Roma, C.; Chicchinelli, N.; Barbieri, A.; Arra, C.; Scognamiglio, G.; Botti, G.; De Luca, A.; Normanno, N. RANTES and IL-6 cooperate in inducing a more aggressive phenotype in breast cancer cells. Oncotarget 2018, 9, 17543–17553. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Tian, L.; Bu, W.; Fan, C.; Gao, X.; Wang, H.; Liao, Y.H.; Li, Y.; Lewis, M.T.; Edwards, D. Wild-type N-Ras, overexpressed in basal-like breast cancer, promotes tumor formation by inducing IL-8 secretion via JAK2 activation. Cell Rep. 2015, 12, 511–524. [Google Scholar] [CrossRef]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2016, 7, 771. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Bouchard, G.; Therriault, H.; Bujold, R.; Saucier, C.; Paquette, B. Induction of interleukin-1β by mouse mammary tumor irradiation promotes triple negative breast cancer cells invasion and metastasis development. Int. J. Radiat. Biol. 2017, 93, 1–10. [Google Scholar] [CrossRef]
- Yamamoto, M.; Taguchi, Y.; Ito-Kureha, T.; Semba, K.; Yamaguchi, N.; Inoue, J.I. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun. 2013, 4, 2299. [Google Scholar] [CrossRef]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Hayden, M.; West, A.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD 4+ T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef]
- Jimi, E.; Strickland, I.; Voll, R.E.; Long, M.; Ghosh, S. Differential role of the transcription factor NF-κB in selection and survival of CD4+ and CD8+ thymocytes. Immunity 2008, 29, 523–537. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Choi, I.-K.; Li, Y.; Oh, E.; Kim, J.; Yun, C.-O. Oncolytic Adenovirus Expressing IL-23 and p35 Elicits IFN-γ- and TNF-α-Co-Producing T Cell-Mediated Antitumor Immunity. PLoS ONE 2013, 8, e67512. [Google Scholar] [CrossRef]
- Gogoi, D.; Chiplunkar, S.V. Targeting gamma delta T cells for cancer immunotherapy: Bench to bedside. Indian J. Med. Res. 2013, 138, 755–761. [Google Scholar]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Sojka, D.K.; Fowell, D.J. Cutting edge: Regulatory T cells selectively attenuate, not terminate, T cell signaling by disrupting NF-κB nuclear accumulation in CD4 T cells. J. Immunol. 2012, 188, 947–951. [Google Scholar] [CrossRef]
- Isomura, I.; Palmer, S.; Grumont, R.J.; Bunting, K.; Hoyne, G.; Wilkinson, N.; Banerjee, A.; Proietto, A.; Gugasyan, R.; Wu, L. C-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 2009, 206, 3001–3014. [Google Scholar] [CrossRef]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-κB transcription factor-dependent, lineage specific transcriptional program promotes regulatory T cell identity and function. Immunity 2017, 47, 450–465. [Google Scholar] [CrossRef]
- Murray, S.E.; Polesso, F.; Rowe, A.M.; Basak, S.; Koguchi, Y.; Toren, K.G.; Hoffmann, A.; Parker, D.C. NF-κB–inducing kinase plays an essential T cell–intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J. Clin. Investig. 2011, 121, 4775–4786. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Papa, S.; Zazzeroni, F.; Pham, C.G.; Bubici, C.; Franzoso, G. Linking JNK signaling to NF-κB: A key to survival. J. Cell Sci. 2004, 117, 5197–5208. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- D’ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef]
- Wang, Q.; Gao, X.; Yu, T.; Yuan, L.; Dai, J.; Wang, W.; Chen, G.; Jiao, C.; Zhou, W.; Huang, Q.; et al. REGγ Controls Hippo Signaling and Reciprocal NF-κB–YAP Regulation to Promote Colon Cancer. Clin. Cancer Res. 2018, 24, 2015–2025. [Google Scholar] [CrossRef]
- Buss, H.; Handschick, K.; Jurrmann, N.; Pekkonen, P.; Beuerlein, K.; Müller, H.; Wait, R.; Saklatvala, J.; Ojala, P.M.; Schmitz, M.L.; et al. Cyclin-Dependent Kinase 6 Phosphorylates NF-κB P65 at Serine 536 and Contributes to the Regulation of Inflammatory Gene Expression. PLoS ONE 2012, 7, e51847. [Google Scholar] [CrossRef]
- Handschick, K.; Beuerlein, K.; Jurida, L.; Bartkuhn, M.; Müller, H.; Soelch, J.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Scharfe, M.; et al. Cyclin-Dependent Kinase 6 Is a Chromatin-Bound Cofactor for NF-κB-Dependent Gene Expression. Mol. Cell 2014, 53, 193–208. [Google Scholar] [CrossRef]
- Aguilar-Quesada, R.; Munoz-Gamez, J.; Martin-Oliva, D.; Peralta-Leal, A.; Quiles-Pérez, R.; Rodriguez-Vargas, J.; De Almodóvar, M.R.; Conde, C.; Ruiz-Extremera, A.; Oliver, F. Modulation of Transcription by PARP-1: Consequences in Carcinogenesis and Inflammation. Curr. Med. Chem. 2007, 14, 1179–1187. [Google Scholar] [CrossRef]
- Senapedis, W.; Crochiere, M.; Rashal, T.; Kashyap, T.; Klebanov, B.; Saint-Martin, J.-R.; Kalid, O.; Shechter, S.; del Alamo, D.; Hamuza, M.; et al. Abstract B198: Selinexor (KPT-330), a novel Selective Inhibitor of Nuclear Export (SINE) shows marked NF-κB inhibition and significant anticancer activity against Non-Small Cell Lung Cancer (NSCLC). Mol. Cancer Ther. 2013, 12, B198. [Google Scholar]
- Kwon, M.; Cho, H.; Lee, Y.; Park, J.; Seo, J. Efficacy of poly(ADP-ribose) polymerase inhibitor olaparib against head and neck cancer cells: Predictions of drug sensitivity based on PAR-p53-NF-κB interactions. Eur. J. Cancer 2016, 69, S120. [Google Scholar] [CrossRef]
- Handa, O.; Yoshida, N.; Fujita, N.; Tanaka, Y.; Ueda, M.; Takagi, T.; Kokura, S.; Naito, Y.; Okanoue, T.; Yoshikawa, T. Molecular mechanisms involved in anti-inflammatory effects of proton pump inhibitors. Inflamm. Res. 2006, 55, 476–480. [Google Scholar] [CrossRef]
- Peng, Y.C.; Huang, L.R.; Shyu, C.L.; Cheng, C.C.; Ho, S.P. Interaction of omeprazole and Helicobacter pylori-induced nuclear factor-κB activation and mediators in gastric epithelial cells. J. Chin. Med Assoc. 2014, 77, 567–572. [Google Scholar] [CrossRef]
- Mondello, P.; Derenzini, E.; Asgari, Z.; Philip, J.; Brea, E.J.; Seshan, V.; Hendrickson, R.C.; de Stanchina, E.; Scheinberg, D.A.; Younes, A. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget 2017, 8, 14017. [Google Scholar] [CrossRef][Green Version]
- Srivastava, R.K.; Kurzrock, R.; Shankar, S. MS-275 Sensitizes TRAIL-Resistant Breast Cancer Cells, Inhibits Angiogenesis and Metastasis, and Reverses Epithelial-Mesenchymal Transition In vivo. Mol. Cancer Ther. 2010, 9, 3254–3266. [Google Scholar] [CrossRef]
- Khong, T.; Sharkey, J.; Spencer, A. The effect of azacitidine on interleukin-6 signaling and nuclear factor-κB activation and its in vitro and in vivo activity against multiple myeloma. Haematologica 2008, 93, 860–869. [Google Scholar] [CrossRef]
- Sullivan, R.; Frederiksen, L.J.; Semenza, G.L.; Graham, C.H.; Paré, G.C. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 2008, 7, 1961–1973. [Google Scholar] [CrossRef]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Nien, C.Y.-C.; Hanna, G.; Rich, J.N.; Provenzale, J.M. Tumor cells upregulate normoxic HIF-1α in response to doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, J.Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. [Google Scholar]
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Badowska-Kozakiewicz, A.M.; Budzik, M.P. Triple-Negative Breast Cancer: Expression of Hypoxia-Inducible Factor 1α in Triple-Negative Breast Cancer with Metastasis to Lymph Nodes. In Breast Cancer and Surgery; INTECHOPEN LIMITED: London, UK, 2018. [Google Scholar]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1–dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Wong, C.C.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Cheng, C.T.; Hou, P.; Lin, Y.P.; Ma, H.; Chung, Y.; Chi, K.; Chen, Y.; Li, W.; Kung, H.J.; et al. HIF-1-alpha links mitochondrial perturbation to the dynamic acquisition of breast cancer tumorigenicity. Oncotarget 2016, 7, 34052–34069. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 regulates the cancer stem cell fate of breast cancer cells in the context of hypoxia and histone deacetylase inhibitors. Sci. Rep. 2019, 9, 9702. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef]
- Shiraishi, A.; Tachi, K.; Essid, N.; Tsuboi, I.; Nagano, M.; Kato, T.; Yamashita, T.; Bando, H.; Hara, H.; Ohneda, O. Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer Sci. 2017, 108, 362–372. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16, 61–70. [Google Scholar] [CrossRef]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.J. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Li, C.; Zhao, Y.; Wu, L.; Du, X.; Han, Z.J.C.P. VEGF-A/Neuropilin 1 pathway confers Cancer Stemness via activating Wnt/β-catenin Axis in breast Cancer cells. Cell. Physiol. Biochem. 2017, 44, 1251–1262. [Google Scholar] [CrossRef]
- Badowska-Kozakiewicz, A.; Sobol, M.; Patera, J. Expression of hypoxia-inducible factor 1α in invasive breast Cancer with metastasis to lymph nodes: Correlation with steroid receptors, HER2 and EPO-R. Adv. Clin. Exp. Med. 2016, 25, 741–750. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, B.; Bao, L.; Jin, L.; Yang, M.; Peng, Y.; Kumar, A.; Wang, J.E.; Wang, C.; Zou, X.; et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Investig. 2018, 128, 1937–1955. [Google Scholar] [CrossRef]
- Milosevic, M.; Chung, P.; Parker, C.; Bristow, R.; Toi, A.; Panzarella, T.; Warde, P.; Catton, C.; Menard, C.; Bayley, A.; et al. Androgen Withdrawal in Patients Reduces Prostate Cancer Hypoxia: Implications for Disease Progression and Radiation Response. Cancer Res. 2007, 67, 6022–6025. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Willard, M.T.; Frederickson, C.E.; Zhong, H.; Simons, J.W. Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3’-kinase/protein kinase B in prostate cancer cells. Clin. Cancer Res. 2003, 9, 2416–2425. [Google Scholar]
- Jia, X.; Hong, Q.; Lei, L.; Li, D.; Li, J.; Mo, M.; Wang, Y.; Shao, Z.; Shen, Z.; Cheng, J.; et al. Basal and therapy-driven hypoxia-inducible factor-1α confers resistance to endocrine therapy in estrogen receptor-positive breast cancer. Oncotarget 2015, 6, 8648–8662. [Google Scholar] [CrossRef]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef]
- Liu, L.P.; Ho, R.L.; Chen, G.G.; Lai, P.B. Sorafenib inhibits hypoxia-inducible factor-1α synthesis: Implications for antiangiogenic activity in hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 5662–5671. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Liang, K.; Pan, T.; Mendelsohn, J.; Fan, Z. Requirement of hypoxia-inducible factor-1α down-regulation in mediating the antitumor activity of the anti–epidermal growth factor receptor monoclonal antibody cetuximab. Mol. Cancer Ther. 2008, 7, 1207–1217. [Google Scholar] [CrossRef]
- Dico, A.L.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef]
- Kelly, C.J.; Hussien, K.; Fokas, E.; Kannan, P.; Shipley, R.J.; Ashton, T.M.; Stratford, M.; Pearson, N.; Muschel, R.J. Regulation of O2 consumption by the PI3K and mTOR pathways contributes to tumor hypoxia. Radiother. Oncol. 2014, 111, 72–80. [Google Scholar] [CrossRef][Green Version]
- Hsu, C.W.; Huang, R.; Khuc, T.; Shou, D.; Bullock, J.; Grooby, S.; Griffin, S.; Zou, C.; Little, A.; Astley, H.; et al. Identification of approved and investigational drugs that inhibit hypoxia-inducible factor-1 signaling. Oncotarget 2016, 7, 8172–8183. [Google Scholar] [CrossRef]
- Williams, S.A.; Anderson, W.C.; Santaguida, M.T.; Dylla, S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Investig. 2013, 93, 970–982. [Google Scholar] [CrossRef]
- Sulaiman, A.; Wang, L. Bridging the divide: Preclinical research discrepancies between triple-negative breast cancer cell lines and patient tumors. Oncotarget 2017, 8, 113269–113281. [Google Scholar] [CrossRef]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17. [Google Scholar] [CrossRef]
- Nugoli, M.; Chuchana, P.; Vendrell, J.; Orsetti, B.; Ursule, L.; Nguyen, C.; Birnbaum, D.; Douzery, E.J.P.; Cohen, P.; Theillet, C. Genetic variability in MCF-7 sublines: Evidence of rapid genomic and RNA expression profile modifications. BMC Cancer 2003, 3, 13. [Google Scholar] [CrossRef]
- Hait, W.N. Anticancer drug development: The grand challenges. Nat. Rev. Drug Discov. 2010, 9, 253–254. [Google Scholar] [CrossRef]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef]
- Ko, A.H.; Murray, J.; Horgan, K.E.; Dauer, J.; Curley, M.; Baum, J.; Louis, C.U.; Lugovskoy, A. A multicenter phase II study of istiratumab (MM-141) plus nab-paclitaxel (A) and gemcitabine (G) in metastatic pancreatic cancer (MPC). J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Adams, S.; Curley, M.D.; Rimkunas, V.; Nie, L.; Tan, G.; Bloom, T.; Iadevaia, S.; Baum, J.; Minx, C.; Czibere, A.; et al. Dual Inhibition of IGF-1R and ErbB3 Enhances the Activity of Gemcitabine and Nab-Paclitaxel in Preclinical Models of Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 2873–2885. [Google Scholar]
- Pace, E.; Adams, S.; Camblin, A.; Curley, M.; Rimkunas, V.; Nie, L.; Iadevaia, S.; Tan, G.; Baum, J.; Czibere, A.G.; et al. Effect of MM-141 on gemcitabine and nab-paclitaxel potentiation in preclinical models of pancreatic cancer through induction of IGF-1R and ErbB3 degradation. J. Clin. Oncol. 2015, 33, 289. [Google Scholar] [CrossRef]
- Baum, J.; Johnson, B.; Adams, S.; Tang, J.; Kohli, N.; Rennard, R.; Sundararajan, P.; Xu, L.; Jiao, Y.; Schoeberl, B.; et al. MM-141, a novel bispecific antibody co-targeting IGF-1R and ErbB3, blocks ligand-induced signaling and demonstrates antitumor activity. Cancer Res. 2012, 72, 2719. [Google Scholar]
- Ledford, H. US cancer institute to overhaul tumour cell lines. Nature 2016, 530, 391. [Google Scholar] [CrossRef]
- Lin, C.H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 16. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Pratt, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-derived Human Breast Cancer Xenograft (PDX) Models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Hasgur, S.; Aryee, K.E.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A. Generation of Immunodeficient Mice Bearing Human Immune Systems by the Engraftment of Hematopoietic Stem Cells. Methods Mol. Biol. 2016, 1438, 67–78. [Google Scholar]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “Humanized” Mice to Study Human Immunity. Curr. Protoc. Immunol. 2008, 81, 15–21. [Google Scholar]
- Rosato, R.R.; Dávila-González, D.; Choi, D.S.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; Dave, B.; Chang, J.C. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res. 2018, 20, 108. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef]


{kind=link}
| Inhibitor | Clinical Trial Number | Mechanism | References |
|---|---|---|---|
| RX-5902 | NCT02003092 | Inhibits phosphorylated p68 RNA helicase preventing nuclear β-catenin translocation and Wnt signaling | [63,64] |
| CB-839 | NCT03057600 | Glutaminase Inhibitor (GSL1). GLS1 has been found to promote stemness via reactive oxygen species/Wnt/ β-catenin signaling | [65] |
| Eribulin mesylate | NCT02513472 | Inhibitor of microtubule dynamics and demonstrated Wnt-related gene suppressive properties | [66] |
| Selinexor | NCT02402764 | Selective inhibitor of nuclear export (SINE) that blocks XPO1 leading to forced nuclear retention of major tumor suppressor proteins reducing β-catenin | [67] |
| Sorafenib | NCT02624700 | Tyrosine protein kinase inhibitor and reduces β-catenin and Wnt signaling | [68] |
| Cetuximab | NCT01097642 | Monoclonal antibody which binds to and inhibits EGFR. Also Inhibits of MAPK which leads to inhibition of β-catenin nuclear activity. | [69] |
| Indomethacin | NCT02950259 | Nonsteroidal anti-inflammatory drug which inhibits prostaglandins which is capable of suppressing β-catenin expression. | [70] |
| Bicalutamide | NCT03090165 | Androgen antagonist preventing Wnt/β-catenin signaling | [71] |
| Inhibitor | Clinical Trial Number | Mechanism | References |
|---|---|---|---|
| Zoledronic Acid | NCT02595138 | Bisphosphonate which inhibits bone resorption and also inhibits farnesyl diphosphate synthase | [89] |
| Erlotinib | NCT02071862 | Epidermal growth factor receptor (EGFR) Inhibitor which can sequester YAP in the cytoplasm | [90] |
| Trametinib | NCT01964924 | MEK1/2 Inhibitor leading to decreased YAP protein levels and transcriptional activity. | [91] |
| Indomethacin | NCT02950259 | Nonsteroidal anti-inflammatory drug that inhibits prostaglandins and is associated with YAP1 stimulation. | [92] |
| Selumetinib (AZD6244) | NCT02583542 | MEK1/2 inhibitor which reduces YAP protein levels | [92] |
| Ipatasertib | NCT02162719 | ATP-competitive, selective AKT inhibitor which can reverse EMT conferred by YAP overexpression | [93] |
| Alisertib (MLN8237) | NCT02187991 | Aurora kinase A inhibitor which was capable of suppressing YAP protein levels | [94] |
| Inhibitor | Clinical Trial Number | Mechanism of Action | References |
|---|---|---|---|
| Ribociclib | NCT03090165 | CDK6 inhibition which prevents CDK6 phosphorylation and activation of NF-κB | [128,129] |
| Veliparib | NCT02032277 | PARP1 and PARP2 inhibitor preventing PARP1 induced NF-κB activity and IL-6/STAT3 expression | [130] |
| Selinexor | NCT02402764 | Selective inhibitor of nuclear export (SINE) that specifically blocks XPO1 leading to forced nuclear retention of major tumor suppressor proteins (TSPs) and inhibits NF-κB transcription. | [131] |
| Reparixin | NCT02370238 | IL8 receptor CXCR1/2 inhibitor | [3] |
| Olaparib | NCT01116648 | PARP Inhibitor which modulates PAR–p53–NF-κB activity | [132] |
| Omeprazol | NCT02950259 | Proton pump inhibitor which interferes with NF-κB activation | [133,134] |
| CUDC-907 | NCT02307240 | PI3K/HDAC inhibitor which was demonstrated to inhibit NF-κB via stimulation IkBα and down-regulation of IKK beta and IRF4 | [135] |
| Entinostat | NCT02708680 | class I HDAC inhibitor which inhibits NF-κB, IL-6 and IL-8 gene signaling | [136] |
| Azacitidine | NCT01349959 | DNA methyltransferase inhibitor, Inhibits IL-6 and NF-κB nuclear translocation | [137] |
| Inhibitor | Clinical Trial Number | Mechanism of Action | References |
|---|---|---|---|
| Bicalutamide | NCT03090165 | Androgen antagonist preventing AR-induced hypoxia signaling | [158,159] |
| Zoledronic Acid | NCT02595138 | Bisphosphonate which inhibits bone resorption and also inhibits HIF-1α transcription via inhibition of RAS/MAPK/ERK1/2 | [160] |
| Eribulin mesylate | NCT02513472 | Inhibitor of microtubule dynamics and can induce tumor vascular remodeling, reducing hypoxia | [66] |
| Everolimus | NCT01931163 | Rapamycin derivative, mTORC1 inhibitor which reduces HIF-1α expression | [161] |
| Sorafenib | NCT02624700 | Tyrosine protein kinase inhibitor and mediated inhibition of HIF-1a and VEGF proteins via modulation of mTOR/p70S6K/4E-BP1 and ERK phosphorylation. | [162] |
| Cetuximab | NCT01097642 | Monoclonal antibody which binds to and inhibits EGFR and down-regulates HIF-1α levels by inhibiting synthesis of HIF-1α. | [163] |
| Trametinib | NCT01964924 | MEK1/2 Inhibitor leading to the inhibition of HIF-1a transcriptional activity | [164] |
| BKM120 | NCT02000882 | P13K/Akt inhibitor which increases mitochondrial oxygen consumption and inhibits hypoxia | [165] |
| Selumetinib (AZD6244) | NCT02583542 | MEK1/2 inhibitor which reduces HIF-1a activity. | [166] |
| Entinostat | NCT02708680 | Class I HDAC inhibitor which inhibits HIF-1α gene signaling | [136] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulaiman, A.; McGarry, S.; Han, X.; Liu, S.; Wang, L. CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers 2019, 11, 1128. https://doi.org/10.3390/cancers11081128
Sulaiman A, McGarry S, Han X, Liu S, Wang L. CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers. 2019; 11(8):1128. https://doi.org/10.3390/cancers11081128
Chicago/Turabian StyleSulaiman, Andrew, Sarah McGarry, Xianghui Han, Sheng Liu, and Lisheng Wang. 2019. "CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs" Cancers 11, no. 8: 1128. https://doi.org/10.3390/cancers11081128
APA StyleSulaiman, A., McGarry, S., Han, X., Liu, S., & Wang, L. (2019). CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers, 11(8), 1128. https://doi.org/10.3390/cancers11081128