The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers?





Abstract
1. Introduction
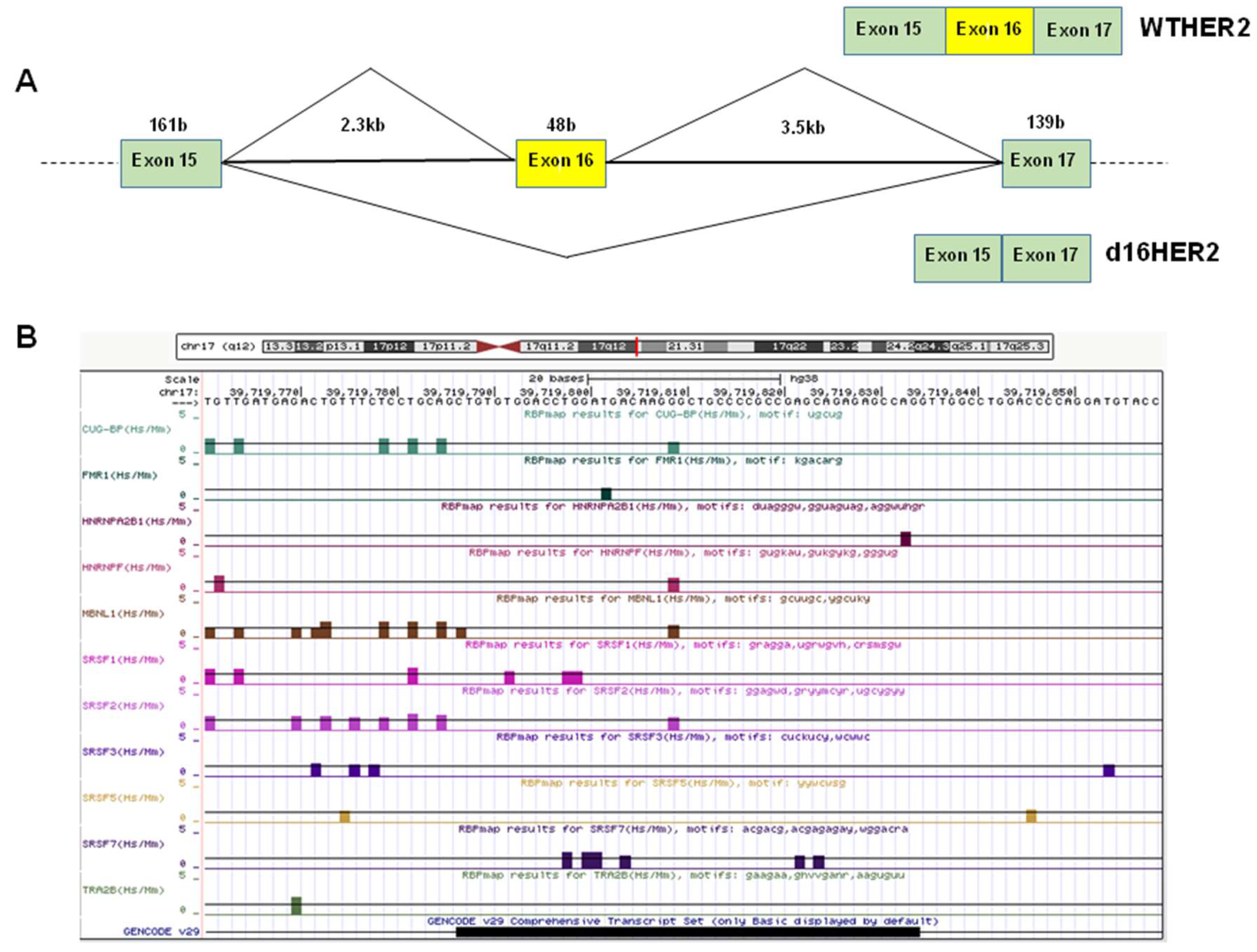
1.1. Alternative Splicing and the Regulation of d16HER2 Expression
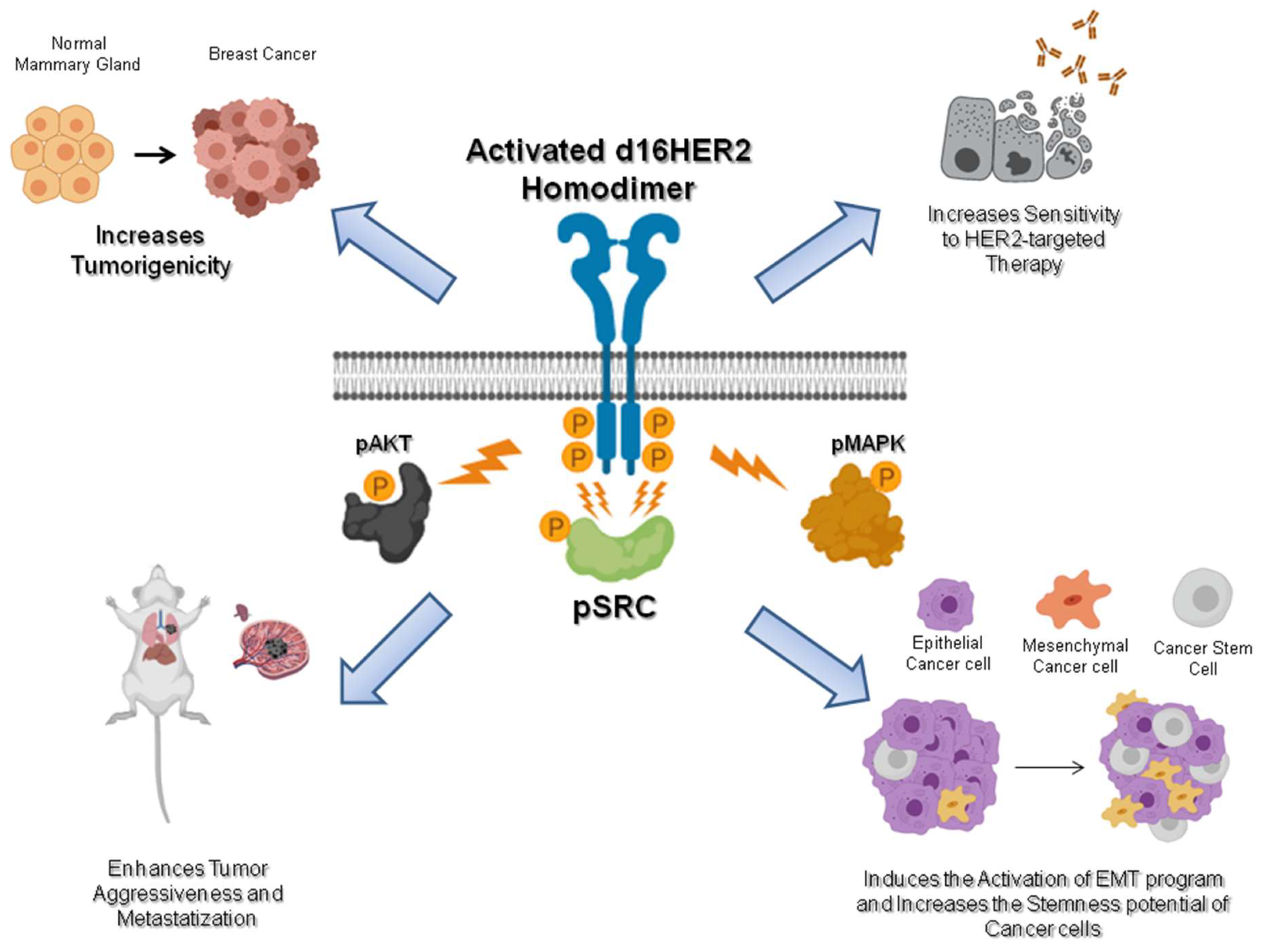
d16HER2: A Crucial Driver of HER2-Driven Tumor Aggressiveness
1.2. d16HER2: The Chief Factor in HER2-Positive Breast Cancer Stem Cells and the EMT Program
1.3. d16HER2: A Promising Predictor of HER2-Targeted Therapy
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coussens, L.; Yang-Feng, T.L.; Liao, Y.C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, F.A.; Schlessinger, J.; Francke, U.; et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Balsari, A.; Campiglio, M.; Pupa, S.M. HER2 as a target for breast cancer therapy. Expert Opin. Biol. Ther. 2010, 10, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef]
- Triulzi, T.; Bianchi, G.V.; Tagliabue, E. Predictive biomarkers in the treatment of HER2-positive breast cancer: An ongoing challenge. Future Oncol. 2016, 12, 1413–1428. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Doherty, J.K.; Bond, C.; Jardim, A.; Adelman, J.P.; Clinton, G.M. The HER-2/neu receptor tyrosine kinase gene encodes a secreted autoinhibitor. Proc. Natl. Acad. Sci. USA 1999, 96, 10869–10874. [Google Scholar] [CrossRef]
- Scott, G.K.; Robles, R.; Park, J.W.; Montgomery, P.A.; Daniel, J.; Holmes, W.E.; Lee, J.; Keller, G.A.; Li, W.L.; Fendly, B.M.; et al. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol. Cell. Biol. 1993, 13, 2247–2257. [Google Scholar] [CrossRef]
- Jackson, C.; Browell, D.; Gautrey, H.; Tyson-Capper, A. Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int. J. Cell. Biol. 2013, 2013, 973584. [Google Scholar] [CrossRef] [PubMed]
- Kwong, K.Y.; Hung, M.C. A novel splice variant of HER2 with increased transformation activity. Mol. Carcinog. 1998, 23, 62–68. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Montiel, N.; Naya-Ruiz, M.; Perez-Santos, M.; Martinez-Contreras, R.D. Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools. Genes 2017, 8, 8100217. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant alternative splicing in breast cancer. J. Mol. Cell. Biol. 2019, 5486617. [Google Scholar] [CrossRef]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef]
- van, R.W.; Le Devedec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de, G.M.; et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar]
- Li, X.H.; Song, J.W.; Liu, J.L.; Wu, S.; Wang, L.S.; Gong, L.Y.; Lin, X. Serine-arginine protein kinase 1 is associated with breast cancer progression and poor patient survival. Med. Oncol. 2014, 31, 83. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Ménard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Brumlik, M.J.; Okamgba, S.U.; Zhu, Y.; Duplessis, T.T.; Parvani, J.G.; Lesko, S.M.; Brogi, E.; Jones, F.E. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer Ther. 2009, 8, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Iezzi, M.; Ghedini, G.C.; Ciravolo, V.; Marzano, G.; Lamolinara, A.; Zappasodi, R.; Gasparini, P.; Campiglio, M.; Amici, A.; et al. Activated d16HER2 homodimers and Src kinase mediate optimal efficacy for trastuzumab. Cancer Res. 2014, 74, 6248–6259. [Google Scholar] [CrossRef]
- Volpi, C.C.; Pietrantonio, F.; Gloghini, A.; Fuca, G.; Giordano, S.; Corso, S.; Pruneri, G.; Antista, M.; Cremolini, C.; Fasano, E.; et al. The landscape of d16HER2 splice variant expression across HER2-positive cancers. Sci. Rep. 2019, 9, 3545. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef] [PubMed]
- Fish, L.; Pencheva, N.; Goodarzi, H.; Tran, H.; Yoshida, M.; Tavazoie, S.F. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 2016, 30, 386–398. [Google Scholar] [CrossRef]
- Watermann, D.O.; Tang, Y.; Zur, H.A.; Jager, M.; Stamm, S.; Stickeler, E. Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006, 66, 4774–4780. [Google Scholar] [CrossRef]
- Slamon, D.J.; Press, M.F.; Godolphin, W.; Ramos, L.; Shek, L.; Stuart, S.G.; Ullrich, A. Studies of the HER2/neu proto-oncogene in human breast cancer. In Cancer Cells; Furth, M., Greaves, M., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989; pp. 371–384. [Google Scholar]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Dankort, D.L.; Hardy, W.R.; Muller, W.J. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol. Cell. Biol. 1994, 14, 7068–7077. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Muller, W.J.; Siegel, P.M. Oncogenic activating mutations in the neu/erbB-2 oncogene are involved in the induction of mammary tumors. Ann. N. Y. Acad. Sci. 1999, 889, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Ryan, E.D.; Cardiff, R.D.; Muller, W.J. Elevated expression of activated forms of Neu/ErbB-2 and Erb-3 are involved in the induction of mammary tumors in transgenic mice: Implications for human breast cancer. EMBO J. 1999, 18, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Muller, W.J. Mutations affecting conserved cysteine residues within the extracellular domain of Neu promote receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 1996, 93, 8878–8883. [Google Scholar] [CrossRef] [PubMed]
- Marchini, C.; Gabrielli, F.; Iezzi, M.; Zanobi, S.; Montani, M.; Pietrella, L.; Kalogris, C.; Rossini, A.; Ciravolo, V.; Castagnoli, L.; et al. The human splice variant delta16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS ONE 2011, 6, e18727. [Google Scholar] [CrossRef] [PubMed]
- Alajati, A.; Sausgruber, N.; Aceto, N.; Duss, S.; Sarret, S.; Voshol, H.; Bonenfant, D.; Tires-Alj, M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013, 73, 5320–5327. [Google Scholar] [CrossRef]
- Turpin, J.; Ling, C.; Crosby, E.J.; Hartman, Z.C.; Simond, A.M.; Chodosh, L.A.; Rennhack, J.P.; Andrechek, E.R.; Ozcelik, J.; Hallett, M.; et al. The ErbB2DeltaEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene 2016, 35, 6053–6064. [Google Scholar] [CrossRef]
- Palladini, A.; Nicoletti, G.; Lamolinara, A.; Dallora, M.; Balboni, T.; Ianzano, M.; Laranga, R.; Landuzzi, L.; Giusti, V.; Ceccarelli, C.; et al. HER2 isoforms co-expression differently tunes mammary tumor phenotypes affecting onset, vasculature and therapeutic response. Oncotarget 2017, 8, 54444–54458. [Google Scholar] [CrossRef]
- Finkle, D.; Quan, Z.R.; Asghari, V.; Kloss, J.; Ghaboosi, N.; Mai, E.; Wong, W.L.; Hollingshead, P.; Schwall, R.; Koeppen, H.; et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin, Cancer Res. 2004, 10, 2499–2511. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.Q.; Alexandrou, A.T.; Li, J.J. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014, 349, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Cancila, V.; Romero-Cordoba, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Volpari, T.; Chiodoni, C.; Hidalgo-Miranda, A.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Korkaya, H.; Wicha, M.S. HER2 and breast cancer stem cells: More than meets the eye. Cancer Res. 2013, 73, 3489–3493. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Ménard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to Trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef]
- Ingthorsson, S.; Andersen, K.; Hilmarsdottir, B.; Maelandsmo, G.M.; Magnusson, M.K.; Gudjonsson, T. HER2 induced EMT and tumorigenicity in breast epithelial progenitor cells is inhibited by coexpression of EGFR. Oncogene 2016, 35, 4244–4255. [Google Scholar] [CrossRef]
- Chen, K.; Dai, X.; Wu, J. Alternative splicing: An important mechanism in stem cell biology. World J. Stem Cells 2015, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nelles, D.A.; Yeo, G.W. Alternative splicing in stem cell self-renewal and diferentiation. Adv. Exp. Med. Biol. 2010, 695, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Warns, J.A.; Davie, J.R.; Dhasarathy, A. Connecting the dots: Chromatin and alternative splicing in EMT. Biochem, Cell. Biol. 2016, 94, 12–25. [Google Scholar] [CrossRef]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Jain, M.V. In vitro tumorigenic assay: Colony forming assay for Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 89–95. [Google Scholar] [CrossRef] [PubMed]
- DiMeo, T.A.; Anderson, K.; Phadke, P.; Fan, C.; Perou, C.M.; Naber, S.; Kuperwasser, C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009, 69, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Ghedini, G.C.; Koschorke, A.; Triulzi, T.; Dugo, M.; Gasparini, P.; Casalini, P.; Palladini, A.; Iezzi, M.; Lamolinara, A.; et al. Pathobiological implications of the d16HER2 splice variant for stemness and aggressiveness of HER2-positive breast cancer. Oncogene 2017, 36, 1721–1732. [Google Scholar] [CrossRef]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intra-tumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 2019, 234, 1768–1779. [Google Scholar] [CrossRef]
- Tilio, M.; Gambini, V.; Wang, J.; Garulli, C.; Kalogris, C.; Andreani, C.; Bartolacci, C.; Elexpuru, Z.M.; Pietrella, L.; Hysi, A.; et al. Irreversible inhibition of Delta16HER2 is necessary to suppress Delta16HER2-positive breast carcinomas resistant to Lapatinib. Cancer Lett. 2016, 381, 76–84. [Google Scholar] [CrossRef]
- Huynh, F.C.; Jones, F.E. MicroRNA-7 inhibits multiple oncogenic pathways to suppress HER2Delta16 mediated breast tumorigenesis and reverse trastuzumab resistance. PLoS ONE 2014, 9, e114419. [Google Scholar] [CrossRef]
- Cittelly, D.M.; Das, P.M.; Salvo, V.A.; Fonseca, J.P.; Burow, M.E.; Jones, F.E. Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis 2010, 31, 2049–2057. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Generation Mechanism | HER2 Isoforms | Cellular Localization |
|---|---|---|
| Proteolytic Cleavage | HER2-ECD (p110) | Soluble extracellular |
| 648-CTF | Anchored in cell membrane | |
| Alternative Splicing | d16HER2 | Transmembrane |
| Herstatin | Soluble extracellular | |
| P100 | Soluble extracellular | |
| Alternative Initiation of translation | 611-CTF (p95HER2) | Transmembrane |
| 687-CTF (p95cyto) | Soluble intracellular | |
| Somatic Mutations | most missense mutations | 20% HER2 extracellular domain |
| duplications/insertions | 80% HER2 transmembrane-extracellular domain |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castagnoli, L.; Ladomery, M.; Tagliabue, E.; Pupa, S.M. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers 2019, 11, 902. https://doi.org/10.3390/cancers11070902
Castagnoli L, Ladomery M, Tagliabue E, Pupa SM. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers. 2019; 11(7):902. https://doi.org/10.3390/cancers11070902
Chicago/Turabian StyleCastagnoli, Lorenzo, Michael Ladomery, Elda Tagliabue, and Serenella M. Pupa. 2019. "The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers?" Cancers 11, no. 7: 902. https://doi.org/10.3390/cancers11070902
APA StyleCastagnoli, L., Ladomery, M., Tagliabue, E., & Pupa, S. M. (2019). The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers, 11(7), 902. https://doi.org/10.3390/cancers11070902