Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia

 , ,
, ,  and
and
Abstract
:1. Introduction
2. HOXA9: A Leukemic Driver in AML
- inhibition of its expression;
- blockade of the specific protein/protein interaction crucial for its mechanism of action;
- or, more specifically as part of a transcription factor, the blockade of the interaction with its cognate sequence on the DNA.
3. Indirect Targeting of HOXA9 at the Expression Level
- DOT1L, which interacts with a large number of fused partners of MLL, themselves present in the non-oncogenic MLL complex;
- menin/LEDGF complex, which interacts with the N-terminal domain of MLL;
- WDR5, which interacts with the C-terminal part of MLL and binds to me-H3K4 marks during the transcription elongation process. Binding of WDR5 in complex with RbBP5 and ASH2L proteins with a WDR5 interacting motif (WIN) changes the conformation of the SET-domain of MLL to activate epigenetic function of the wild-type MLL complex;
- pTEFb complex, which makes contact with both MLL and DOT1L proteins and traps BRD4 to a large MLL-fusion complex.
- MOF, which binds to the SET domain in the C-terminal part of MLL protein and acetylates H4K16 position at transcriptional initiation stage [121];
- UBE2O, which promotes wild-type MLL degradation in response to IL1 treatment and subsequent IRAK4 (interleukin-1 receptor-associated kinase 4) activation pathway [122] in order to remove the WT-MLL complex from the chromatin and thus to favor oncogenic MLL chimera complex binding to the chromatin.
3.1. Targeting MLL-Interacting Partners at the Protein/Protein Interaction Level
3.1.1. Targeting Disruptor of Telomeric Silencing 1-like (DOT1L)/Mixed Lineage Leukemia (MLL) Proteins Interaction
- the pyrimidyl-aminoquinoline derivative 9e, that interacts with DOT1L at the µM range and reduces HOXA9 and MEIS1 expression [139];
- compounds 3 and 9, identified by high throughput screening [140];
- DC_L115, which binds DOT1L at sub-micromolar concentration [141];
- 2-chloro benzothiophene derivative 12 and aza-benzimidazole derivative 13, as two orally bioavailable DOT1L inhibitors [142];
- the phenoxyacetamide derivatives L01, L03, L04, and L05, identified by hierarchical docking-based virtual screening and molecular dynamic simulation [143];
- and massonianoside B, identified by use of a pharmacophore-based in silico screening [144].
3.1.2. Targeting the Menin/Mixed Lineage Leukemia (MLL) Proteins Interaction Interface
3.1.3. Targeting WDR5/MLL Interaction
3.1.4. Targeting Cyclin-Dependent Kinase 9 (CDK9)
3.1.5. Targeting Mixed Lineage Leukemia (MLL) Protein Degradation
3.1.6. Targeting Other Epigenetic Factors Associated with the MLL Complex
3.2. Targeting HOXA9 Expression Through MLL-Independent Epigenetic Modifiers
4. Direct Targeting of HOXA9
4.1. Direct Targeting of HOXA9 Using Inhibitors of HOXA9/Cofactor Interaction
4.2. Direct Targeting of HOXA9/DNA Interaction Using Sequence-Selective DNA Ligands
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| ATAC-seq | assay for transposase-accessible chromatin with high throughput sequencing |
| ATM | ataxia telangiectasia mutated |
| ATR | ATM and RAD3-related |
| ATRA | all-trans retinoic acid |
| BET-seq | binding energy topography by sequencing |
| BiFC | bimolecular fluorescence complementation |
| BMP | bone morphogenic protein |
| BRD4 | bromodomain-related protein 4 |
| CBP | CREB-binding protein |
| CDK9 | cyclin-dependent kinase 9 |
| CFU-G | colony forming unit-granulocyte |
| CFU-GM | colony forming unit-granulocyte and monocyte |
| CFU-M | colony forming unit-monocyte |
| ChIA-Pet | chromatin interaction analysis by paired-end tag |
| ChIP | chromatine immune-precipitation CKII, casein kinase II |
| CTCF | CCCTC-binding factor |
| CUT&RUN | cleavage under targets and release using nuclease |
| D-2-HG | D-2-hydroxyglutarate |
| DHODH | dihydroorotate dehydrogenase |
| DNase-Seq | deoxyribonuclease I (DNase I)-hypersensitive site with high throughput sequencing |
| DNMT | DNA methyl transferase |
| DOT1L | disruptor of telomeric silencing 1-like protein |
| Erg | ETS-related gene |
| EVI1 | ecotopic viral integration site 1 |
| FLT3 | Fms-like tyrosine kinase 3 |
| GFI1 | growth factor independent 1 |
| GSEA | gene set enrichment analysis |
| HAT | histone acetyl-transferase |
| HD | homeodomain |
| HDAC | histone deacetylase |
| HEXIM1 | hexamethylene bisacetamide (HMBA) inducible protein 1 |
| HMT | histone methyltransferase |
| HOXA9 | homeobox A9 |
| IDH | isocitrate dehydrogenase |
| IL1B | interleukin 1 beta |
| IRAK4 | interleukin-1 receptor-associated kinase 4 |
| KDM4C | lysine-specific demethylase 4C |
| LEDGF | lens epithelium-derived growth factor |
| Lmo2 | LIM domain only 2 |
| LSD1 | lysine-specific demethylase 1 |
| MAPK | mitogen-activated protein kinase |
| MEF2C | myocyte enhancer factor 2C |
| MEIS | myeloid ecotropic viral integration site |
| MLL | mixed lineage leukemia |
| MOF | males absent on the first |
| MPAL | mixed-phenotype associated lineage leukemia |
| Myb | myeloblastosis gene |
| NPM1 | nucleophosmin 1 |
| NSD1 | nuclear receptor binding SET domain protein 1 |
| NUP98 | nucleoporin 98kDa |
| OPN | osteopontin |
| PBX | pre-B-cell leukemia transcription factor |
| PKC | protein kinase C |
| PRC2 | polycomb repressive complex 2 |
| PRMT1 | protein arginine N-methyltransferase 1 |
| PTD | partial tandem duplication |
| pTEFb | positive transcription elongation factor b |
| RIME | immunoprecipitation-coupled mass spectrometry of endogenous protein |
| SAM | S-adenosyl-methionine |
| SELEX-seq | systematic evolution of ligands by exponential enrichment followed by sequencing |
| shRNA | short non-coding RNA |
| SMAD4 | mothers against decapentaplegic homolog 4 |
| TDG | thymine DNA glycosylase |
| TGFβ | transforming growth factor beta |
| WDR5 | WD repeat protein 5 |
| WT | wild-type |
| XPO-1 | exportin-1 |
| 4C-seq | circularized chromosome conformation capture with high throughput sequencing |
References
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Akhtar, A.; Luscombe, N.M. Large-Scale Nuclear Architecture and Transcriptional Control. Subcell. Biochem. 2011, 52, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Gehring, W.J.; Affolter, M.; Burglin, T. Homeodomain Proteins—Annual Review of Biochemistry. Annu. Rev. Biochem. 1994, 63, 6487–6526. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Sauvageau, G.; Humphries, R.K.; Largman, C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 1996, 14, 281–291. [Google Scholar] [CrossRef]
- Van Oostveen, J.W.; Bijl, J.J.; Raaphorst, F.M.; Walboomers, J.J.M.; Meijer, C.J.L.M. The role of Homeobox genes in normal hematopoiesis and hematological malignancies. Leukemia 1999, 13, 1675–1690. [Google Scholar] [CrossRef] [Green Version]
- Argiropoulos, B.; Humphries, R.K. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–6776. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, R.A.; Pandha, H.S.; Simpson, G.R.; Pettengell, R.; Poterlowicz, K.; Thompson, A.; Harrington, K.; El-Tanani, M.; Morgan, R. Inhibition of HOX/PBX dimer formation leads to necroptosis in acute myeloid leukemia cells. Oncotarget 2017, 8, 89566–89579. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Helms, J.A.; Chang, H.Y. Regeneration, repair and remembering identity: The three Rs of Hox gene expression. Trends Cell Biol. 2009, 19, 268–275. [Google Scholar] [CrossRef]
- Kachgal, S.; Mace, K.A.; Boudreau, N.J. The dual roles of homeobox genes in vascularization and wound healing. Cell Adhes. Migr. 2012, 6, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.S. The Role of HOX Genes in the Development and Function of the Female Reproductive Tract. Semin. Reprod. Med. 2002, 18, 081–090. [Google Scholar] [CrossRef]
- Akbas, G.E.; Taylor, H.S. HOXC and HOXD Gene Expression in Human Endometrium: Lack of Redundancy with HOXA Paralogs1. Biol. Reprod. 2004, 70, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, I.A.; Acerini, C.L. Factors controlling testis descent. Eur. J. Endocrinol. 2008, 159, S75–S82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raines, A.M.; Adam, M.; Magella, B.; Meyer, S.E.; Grimes, H.L.; Dey, S.K.; Potter, S.S. Recombineering-based dissection of flanking and paralogous Hox gene functions in mouse reproductive tracts. Development 2013, 140, 2942–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Taylor, H.S. The role of hox genes in female reproductive tract development, adult function, and fertility. Cold Spring Harb. Perspect. Med. 2016, 6, 023002. [Google Scholar] [CrossRef] [PubMed]
- Awgulewitsch, A. Hox in hair growth and development. Naturwissenschaften 2003, 90, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Freschi, G.; Taddei, A.; Bechi, P.; Faiella, A.; Gulisano, M.; Cillo, C.; Bucciarelli, G.; Boncinelli, E. Expression of HOX homeobox genes in the adult human colonic mucosa (and colorectal cancer?). Int. J. Mol. Med. 2005, 16, 581–587. [Google Scholar]
- Mahdipour, E.; Mace, K.A. Hox transcription factor regulation of adult bone-marrow-derived cell behaviour during tissue repair and regeneration. Expert Opin. Biol. Ther. 2011, 11, 1079–1090. [Google Scholar] [CrossRef]
- Rux, D.R.; Wellik, D.M. Hox genes in the adult skeleton: Novel functions beyond embryonic development. Dev. Dyn. 2017, 246, 310–317. [Google Scholar] [CrossRef]
- Dunwell, T.L.; Holland, P.W.H. Diversity of human and mouse homeobox gene expression in development and adult tissues. BMC Dev. Biol. 2016, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 2002, 30, 49–57. [Google Scholar] [CrossRef]
- Barba, P.; Magli, M.C.; Tiberio, C.C.C. HOX gene expression in human cancers. Adv. Exp. Med. Biol. 1993, 348, 45–57. [Google Scholar] [PubMed]
- Cillo, C. HOX genes in human cancers. Invasion Metastasis 1994, 14, 38–49. [Google Scholar] [PubMed]
- Goodman, F.R.; Scambler, P.J. Human HOX gene mutations. Clin. Genet. 2001, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cantile, M.; Franco, R.; Schiavo, G.; Procino, A.; Cindolo, L.; Botti, G. The HOX genes network in uro-genital cancers: Mechanisms and potential therapeutic implications. Curr. Med. Chem. 2011, 18, 4872–4884. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; El-Tanani, M. HOX Genes as Potential Markers of Circulating Tumour Cells. Curr. Mol. Med. 2016, 16, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Huang, Q.; Wei, G.-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528. [Google Scholar] [CrossRef]
- Li, Z.; Chen, P.; Su, R.; Hu, C.; Li, Y.; Elkahloun, A.G.; Zuo, Z.; Gurbuxani, S.; Arnovitz, S.; Weng, H.; et al. PBX3 and MEIS1 Cooperate in Hematopoietic Cells to Drive Acute Myeloid Leukemias Characterized by a Core Transcriptome of the MLL-Rearranged Disease. Cancer Res. 2016, 76, 619–629. [Google Scholar] [CrossRef]
- Nakamura, T.; Largaespada, D.A.; Lee, M.P.; Johnson, L.A.; Ohyashiki, K.; Toyama, K.; Chen, S.J.; Willman, C.L.; Chen, I.M.; Feinberg, A.P.; et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat. Genet. 1996, 12, 154–158. [Google Scholar] [CrossRef]
- Kroon, E.; Krosl, J.; Thorsteinsdottir, U.; Baban, S.; Buchberg, A.M.; Sauvageau, G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998, 17, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Borrow, J.; Shearman, A.M.; Stanton, V.P.; Becher, R.; Collins, T.; Williams, A.J.; Dubé, I.; Katz, F.; Kwong, Y.L.; Morris, C.; et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat. Genet. 1996, 12, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Rio-Machin, A.; Gómez-López, G.; Muñoz, J.; Garcia-Martinez, F.; Maiques-Diaz, A.; Alvarez, S.; Salgado, R.N.; Shrestha, M.; Torres-Ruiz, R.; Haferlach, C.; et al. The molecular pathogenesis of the NUP98-HOXA9 fusion protein in acute myeloid leukemia. Leukemia 2017, 32, 2000–2005. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Brindle, P.K.; Schnabel, C.A.; Pritchard, C.E.J.; Cleary, M.L.; van Deursen, J.M.A. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 1999, 19, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Thorsteinsdottir, U.; Mayotte, N.; Nakamura, T.; Sauvageau, G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001, 20, 350–361. [Google Scholar] [CrossRef]
- Zhang, Y.; Morrone, G.; Zhang, J.; Chen, X.; Lu, X.; Ma, L.; Moore, M.; Zhou, P. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J. 2003, 22, 6057–6067. [Google Scholar] [CrossRef] [Green Version]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef]
- Debernardi, S.; Lillington, D.M.; Chaplin, T.; Tomlinson, S.; Amess, J.; Rohatiner, A.; Lister, T.A.; Young, B.D. Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosom. 2003, 37, 149–158. [Google Scholar] [CrossRef]
- Andreeff, M.; Ruvolo, V.; Gadgil, S.; Zeng, C.; Coombes, K.; Chen, W.; Kornblau, S.; Barón, A.E.; Drabkin, H.A. HOX expression patterns identify a common signature for favorable AML. Leukemia 2008, 22, 2041–2047. [Google Scholar] [CrossRef] [Green Version]
- Braekeleer, E.D.; Douet-Guilbert, N.; Basinko, A.; Bris, M.J.L.; Morel, F.; De Braekeleer, M. Hox gene dysregulation in acute myeloid leukemia. Futur. Oncol. 2014, 10, 475–495. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Sun, J.; Liu, F.; Zhang, H.; Ma, Y. Higher expression levels of the HOXA9 gene, closely associated with MLL-PTD and EZH2 mutations, predict inferior outcome in acute myeloid leukemia. Onco. Targets Ther. 2016, 9, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawagoe, H.; Humphries, R.K.; Blair, A.; Sutherland, H.J.; Hogge, D.D. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 1999, 13, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozovskaia, T.; Feinstein, E.; Mor, O.; Foa, R.; Blechman, J.; Nakamura, T.; Croce, C.M.; Cimino, G.; Canaani, E. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4: 11) abnormality. Oncogene 2001, 20, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Armstrong, S.A.; Neuberg, D.S.; Sallan, S.E.; Silverman, L.B.; Korsmeyer, S.J.; Look, A.T. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: Dominance of HOX dysregulation. Blood 2003, 102, 262–268. [Google Scholar] [CrossRef]
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003, 17, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, M.; Tiacci, E.; Bergomas, R.; Bigerna, B.; Venturini, E.; Minardi, S.P.; Meani, N.; Diverio, D.; Bernard, L.; Tizzoni, L.; et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 2005, 106, 899–902. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Kennedy, A.; Zhou, X.; Radtke, I.; Phillips, L.A.; Shurtleff, S.A.; Downing, J.R. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 2007, 21, 2000–2009. [Google Scholar] [CrossRef]
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. IDH2 and NPM1 mutations cooperate to activate Hoxa9/Meis1 and Hypoxia pathways in acute myeloid Leukemia. Cancer Res. 2015, 75, 2005–2016. [Google Scholar] [CrossRef]
- Slany, R.K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, R. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 2011, 152, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Hess, J.L. The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis. 2011, 7, 283–301. [Google Scholar] [CrossRef]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.J.; Hess, J.L. The PAF Complex Synergizes with MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Rossetti, G.; Arnesano, F.; Ippoliti, E.; Natile, G.; Carloni, P. Molecular recognition of platinated DNA from chromosomal HMGB1. J. Chem. Theory Comput. 2014, 10, 3578–3584. [Google Scholar] [CrossRef]
- Okuda, H.; Stanojevic, B.; Kanai, A.; Kawamura, T.; Takahashi, S.; Matsui, H.; Takaori-Kondo, A.; Yokoyama, A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Investig. 2017, 127, 1918–1931. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a Cyclin-Dependent Kinase Controlling Elongation by RNA Polymerase II. Mol. Cell. Biol. 2002, 20, 2629–2634. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, A.T.; Huang, J.; Lei, M.; Hua, X. Menin as a hub controlling mixed lineage leukemia. BioEssays 2012, 34, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C.; Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; et al. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.; Katryniok, C.; Scholz, B.; Merkens, J.; Löscher, D.; Marschalek, R.; Steinhilber, D. Inhibition of class I HDACs abrogates the dominant effect of MLL-AF4 by activation of wild-type MLL. Oncogenesis 2014, 3, e127. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.J.; Korsmeyer, S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 1995, 378, 505–508. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- He, M.; Chen, P.; Arnovitz, S.; Li, Y.; Huang, H.; Neilly, M.B.; Wei, M.; Rowley, J.D.; Chen, J.; Li, Z. Two isoforms of HOXA9 function differently but work synergistically in human MLL-rearranged leukemia. Blood Cells Mol. Dis. 2012, 49, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012, 120, 1130–1136. [Google Scholar] [CrossRef]
- Whitman, S.P.; Liu, S.; Vukosavljevic, T.; Rush, L.J.; Yu, L.; Liu, C.; Klisovic, M.I.; Maharry, K.; Guimond, M.; Strout, M.P.; et al. The MLL partial tandem duplication: Evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005, 106, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Kühn, M.W.M.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, C.; Zhao, J.; Zhu, Y.; Weng, X.; Chen, Q.; Sun, H.; Mi, J.Q.; Li, J.; Zhu, J.; et al. Inactivation of PBX3 and HOXA9 by down-regulating H3K79 methylation represses NPM1-mutated leukemic cell survival. Theranostics 2018, 8, 4359–4371. [Google Scholar] [CrossRef]
- Gurumurthy, M.; Tan, C.H.; Ng, R.; Zeiger, L.; Lau, J.; Lee, J.; Dey, A.; Philp, R.; Li, Q.; Lim, T.M.; et al. Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J. Mol. Biol. 2008, 378, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Monroe, S.C.; Jo, S.Y.; Sanders, D.S.; Basrur, V.; Elenitoba-Johnson, K.S.; Slany, R.K.; Hess, J.L. MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp. Hematol. 2011, 39, 77–86. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef]
- Gu, X.; Ebrahem, Q.; Mahfouz, R.Z.; Hasipek, M.; Enane, F.; Radivoyevitch, T.; Rapin, N.; Przychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Investig. 2018, 128, 4260–4279. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Kok, C.H.; Brown, A.L.; Ekert, P.G.; D’Andrea, R.J. Gene expression analysis reveals HOX gene upregulation in trisomy 8 AML. Leukemia 2010, 24, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, J.L.; Lopez Martí, J.M.; Kumar, A.; Kontro, M.; Edgren, H.; Parsons, A.; Lundán, T.; Wolf, M.; Porkka, K.; Heckman, C.A. Chimeric NUP98–NSD1 transcripts from the cryptic t(5;11)(q35.2;p15.4) in adult de novo acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, G.; Takeda, A.; Camarata, T.; Moore, M.A.; Viale, A.; Yaseen, N.R. The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J. Biol. Chem. 2004, 279, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, L.; Pineault, N.; Wasslavik, C.; Humphries, R.K. Candidate genes for expansion and transformation of hematopoietic stem cells by NUP98-HOX fusion genes. PLoS ONE 2007, 2, e768. [Google Scholar] [CrossRef] [PubMed]
- Taketani, T.; Taki, T.; Shibuya, N.; Kikuchi, A.; Hanada, R.; Hayashi, Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13). Cancer Res. 2002, 62, 4571–4574. [Google Scholar] [PubMed]
- Taketani, T.; Taki, T.; Shibuya, N.; Hayashi, Y.; Taketani, T.; Ito, E.; Kitazawa, J.; Terui, K. The HOXD11 gene is fused to the NUP98 gene in acute myeloid leukemia with t(2;11)(q31;p15). Cancer Res. 2002, 62, 33–37. [Google Scholar] [PubMed]
- Jankovic, D.; Gorello, P.; Liu, T.; Ehret, S.; La Starza, R.; Desjobert, C.; Baty, F.; Brutsche, M.; Jayaraman, P.S.; Santoro, A.; et al. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 2008, 111, 5672–5682. [Google Scholar] [CrossRef] [PubMed]
- Camós, M.; Esteve, J.; Jares, P.; Colomer, D.; Rozman, M.; Villamor, N.; Costa, D.; Carrió, A.; Nomdedéu, J.; Montserrat, E.; et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 2006, 66, 6947–6954. [Google Scholar] [CrossRef]
- Jin, G.; Yamazaki, Y.; Takuwa, M.; Takahara, T.; Kaneko, K.; Kuwata, T.; Miyata, S.; Nakamura, T. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood 2007, 109, 3998–4005. [Google Scholar] [CrossRef]
- Chase, A.; Reiter, A.; Burci, L.; Cazzaniga, G.; Biondi, A.; Pickard, J.; Roberts, I.A.; Goldman, J.M.; Cross, N.C. Fusion of ETV6 to the caudal-related homeobox gene CDX2 in acute myeloid leukemia with the t(12;13)(p13;q12). Blood 1999, 93, 1025–1031. [Google Scholar]
- Novak, R.L.; Harper, D.P.; Caudell, D.; Slape, C.; Beachy, S.H.; Aplan, P.D. Gene expression profiling and candidate gene resequencing identifies pathways and mutations important for malignant transformation caused by leukemogenic fusion genes. Exp. Hematol. 2012, 40, 1016–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vlierberghe, P.; Van Grotel, M.; Tchinda, J.; Lee, C.; Beverloo, H.B.; Van Der Spek, P.J.; Stubbs, A.; Cools, J.; Nagata, K.; Fornerod, M.; et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 2008, 111, 4668–4680. [Google Scholar] [CrossRef] [PubMed]
- Dumezy, F.; Preudhomme, C.; Nibourel, O.; Labis, E.; Renneville, A.; Mayeur-Rousse, C. Acute myeloid leukemia with translocation t(3;5): New molecular insights. Haematologica 2013, 98, e52–e54. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.; Choi, J.R.; Kim, M.J.; Kim, S.Y.; Lee, H.J.; Suh, J.T.; Yoon, H.J.; Lee, J.; Lee, S.; Lee, W.I.; et al. Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: Clinical and laboratory study with review of the literature. Cancer Genet. Cytogenet. 2010, 199, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Koya, J.; Kataoka, K.; Sato, T.; Bando, M.; Kato, Y.; Tsuruta-Kishino, T.; Kobayashi, H.; Narukawa, K.; Miyoshi, H.; Shirahige, K.; et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat. Commun. 2016, 7, 10924. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hönes, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for GFI136N-associated murine/human AML. Exp. Hematol. 2016, 44, 713–726. [Google Scholar] [CrossRef]
- Chen, S.; Yu, J.; Lv, X.; Zhang, L. HOXA9 is critical in the proliferation, differentiation, and malignancy of leukaemia cells both in vitro and in vivo. Cell Biochem. Funct. 2017, 35, 433–440. [Google Scholar] [CrossRef]
- Li, A.; Yang, Y.; Gao, C.; Lu, J.; Jeong, H.W.; Liu, B.H.; Tang, P.; Yao, X.; Neuberg, D.; Huang, G.; et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J. Clin. Investig. 2013, 123, 4195–4207. [Google Scholar] [CrossRef]
- Dickson, G.; Liberante, F.; Kettyle, L.; O’Hagan, K.; Finnegan, D.; Bullinger, L.; Geerts, D.; McMullin, M.F.; Lappin, T.; Mills, K.; et al. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica 2013, 98, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Lambert Jambon, S.; Depauw, S.; Bouhlel, M.A.; Figeac, M.; David-Cordonnier, M.-H. HOXA9 transcription factor as a target in acute myeloid leukemia: Transcription, cellular and in vivo consequences of its invalidation. Eur. J. Cancer 2016, 69, S23. [Google Scholar] [CrossRef]
- Breitinger, C.; Maethner, E.; Garcia-Cuellar, M.P.; Slany, R.K. The homeodomain region controls the phenotype of HOX-induced murine leukemia. Blood 2012, 120, 4018–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, K.R.; Sykes, D.B.; Pasillas, M.; Kamps, M.P. Hoxa9 Immortalizes a Granulocyte-Macrophage Colony-Stimulating Factor-Dependent Promyelocyte Capable of Biphenotypic Differentiation to Neutrophils or Macrophages, Independent of Enforced Meis Expression. Mol. Cell. Biol. 2002, 20, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Passegué, E.; Fong, S.; Largman, C.; Lawrence, H.J. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood 2007, 109, 4732–4738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, C.R.; Vegi, N.; Mulaw, M.A.; Edmaier, K.E.; Rawat, V.P.S.; Dolnik, A.; Bullinger, L.; Heilmeier, B.; Quintanilla-Fend, L.; Spiekermann, K.; et al. The leukemogenicity of Hoxa9 depends on alternative splicing. Leukemia 2014, 28, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Dintilhac, A.; Bihan, R.; Guerrier, D.; Deschamps, S.; Pellerin, I. A conserved non-homeodomain Hoxa9 isoform interacting with CBP is co-expressed with the “typical” Hoxa9 protein during embryogenesis. Gene Expr. Patterns 2004, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Vijapurkar, U.; Fischbach, N.; Shen, W.; Brandts, C.; Stokoe, D.; Lawrence, H.J.; Largman, C. Protein kinase C-mediated phosphorylation of the leukemia-associated HOXA9 protein impairs its DNA binding ability and induces myeloid differentiation. Mol. Cell. Biol. 2004, 24, 3827–3837. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sitwala, K.; Bronstein, J.; Sanders, D.; Dandekar, M.; Collins, C.; Robertson, G.; MacDonald, J.; Cezard, T.; Bilenky, M.; et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood 2012, 119, 388–398. [Google Scholar] [CrossRef]
- Zhong, X.; Prinz, A.; Steger, J.; Garcia-Cuellar, M.P.; Radsak, M.; Bentaher, A.; Slany, R.K. HoxA9 transforms murine myeloid cells by a feedback loop driving expression of key oncogenes and cell cycle control genes. Blood Adv. 2018, 2, 3137–3148. [Google Scholar] [CrossRef]
- Dorsam, S.T.; Ferrell, C.M.; Dorsam, G.P.; Derynck, M.K.; Vijapurkar, U.; Khodabakhsh, D.; Pau, B.; Bernstein, H.; Haqq, C.M.; Largman, C.; et al. The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood 2004, 103, 1676–1684. [Google Scholar] [CrossRef] [Green Version]
- Calero-Nieto, F.J.; Joshi, A.; Bonadies, N.; Kinston, S.; Chan, W.-I.; Gudgin, E.; Pridans, C.; Landry, J.-R.; Kikuchi, J.; Huntly, B.J.; et al. HOX-mediated LMO2 expression in embryonic mesoderm is recapitulated in acute leukaemias. Oncogene 2013, 32, 5471–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, C.A.; Bei, L.; Wang, H.; Platanias, L.C.; Eklund, E.A. HoxA10 Protein Regulates Transcription of Gene Encoding Fibroblast Growth Factor 2 (FGF2) in Myeloid Cells. J. Biol. Chem. 2012, 287, 18230–18248. [Google Scholar] [CrossRef] [PubMed]
- Steger, J.; Füller, E.; Garcia-Cuellar, M.-P.; Hetzner, K.; Slany, R.K. Insulin-like growth factor 1 is a direct HOXA9 target important for hematopoietic transformation. Leukemia 2015, 29, 901–908. [Google Scholar] [CrossRef]
- Brumatti, G.; Salmanidis, M.; Kok, C.H.; Bilardi, R.A.; Sandow, J.J.; Silke, N.; Manson, K.; Visser, J.; Jabbour, A.M.; Glaser, S.P.; et al. HoxA9 regulated Bcl-2 expression mediates survival of myeloid progenitors and the severity of HoxA9-dependent leukemia. Oncotarget 2013, 4, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Kantarjian, H.; Estey, E.; Manshouri, T.; Chan, C.; Rahman, E.A.; Kornblau, S.; Cortes, J.; Thomas, D.; Pierce, S.; et al. The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Hess, J.L.; Bittner, C.B.; Zeisig, D.T.; Bach, C.; Fuchs, U.; Borkhardt, A.; Frampton, J.; Slany, R.K. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006, 108, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhou, B.; Armstrong, S.A.; Dou, Y.; Hess Correspondence, J.L.; Mao, F.; Xu, J.; Miao, H.; Zou, Z.; Tran, L.; et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis In Brief Cancer Cell HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell 2018, 34. [Google Scholar] [CrossRef]
- Moskow, J.J.; Bullrich, F.; Huebner, K.; Daar, I.O.; Buchberg, A.M. Meis1, a PBX1-related homeobox gene involved in myeloid leukemia in BXH-2 mice. Mol. Cell. Biol. 1995, 15, 5434–5443. [Google Scholar] [CrossRef] [Green Version]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 2001, 21, 224–234. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical Association and Coordinate Function of the H3 K4 Methyltransferase MLL1 and the H4 K16 Acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, K.; Volk, A.G.; Haug, J.S.; Marshall, S.A.; Woodfin, A.R.; Bartom, E.T.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Sullivan, K.D.; et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell 2017, 168, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xia, X.; Reisenauer, M.R.; Hemenway, C.S.; Kone, B.C. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J. Biol. Chem. 2006, 281, 18059–18068. [Google Scholar] [CrossRef]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Basavapathruni, A.; Jin, L.; Daigle, S.R.; Majer, C.R.A.; Therkelsen, C.A.; Wigle, T.J.; Kuntz, K.W.; Chesworth, R.; Pollock, R.M.; Scott, M.P.; et al. Conformational Adaptation Drives Potent, Selective and Durable Inhibition of the Human Protein Methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80, 971–980. [Google Scholar] [CrossRef]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deshpande, A.J.; Banka, D.; Bernt, K.M.; Dias, S.; Buske, C.; Olhava, E.J.; Daigle, S.R.; Richon, V.M.; Pollock, R.M.; et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 2013, 27, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Rau, R.E.; Rodriguez, B.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 684225. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ge, W. The histone methyltransferase DOT1L inhibits osteoclastogenesis and protects against osteoporosis. Cell Death Dis. 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant- IDH2 acute myeloid leukemia treated with enasidenib. Blood 2018, 133, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L Inhibitor EPZ-5676 Displays Synergistic Antiproliferative Activity in Combination with Standard of Care Drugs and Hypomethylating Agents in MLL-Rearranged Leukemia Cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef]
- Kühn, M.W.M.; Hadler, M.J.; Daigle, S.R.; Koche, R.P.; Krivtsov, A.V.; Olhava, E.J.; Caligiuri, M.A.; Huang, G.; Bradner, J.E.; Pollock, R.M.; et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica 2015, 100, e190–e193. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Gong, Z.; Chen, J.; Kim, J.E. Characterization of the DOT1L network: Implications of diverse roles for DOT1L. Protein J. 2010, 29, 213–223. [Google Scholar] [CrossRef]
- Liu, W.; Deng, L.; Song, Y.; Redell, M. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS ONE 2014, 9, e98270. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Y.; Liu, N.; Li, L.; Xiao, S.; Li, X.; Chen, K.; Luo, C.; Chen, S.; Chen, H. Design, synthesis and anti leukemia cells proliferation activities of pyrimidylaminoquinoline derivatives as DOT1L inhibitors. Bioorg. Chem. 2018, 80, 649–654. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Chen, Y.; Liu, J.; Xiao, S.; Lian, F.; Zhang, N.; Ding, H.; Zhang, Y.; Chen, K.; et al. Discovery of potent DOT1L inhibitors by AlphaLISA based High Throughput Screening assay. Bioorg. Med. Chem. 2018, 26, 1751–1758. [Google Scholar] [CrossRef]
- Chen, S.; Li, L.; Chen, Y.; Hu, J.; Liu, J.; Liu, Y.C.; Liu, R.; Zhang, Y.; Meng, F.; Zhu, K.; et al. Identification of Novel Disruptor of Telomeric Silencing 1-like (DOT1L) Inhibitors through Structure-Based Virtual Screening and Biological Assays. J. Chem. Inf. Model. 2016, 56, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, H.; Stauffer, F.; Caravatti, G.; Vollmer, S.; Machauer, R.; Holzer, P.; Möbitz, H.; Scheufler, C.; Klumpp, M.; et al. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Wang, H.; Zou, Y.; Zhang, S.; Xiao, J.; Jiang, G.; Zhang, Y.; Lai, Y. Identification of phenoxyacetamide derivatives as novel DOT1L inhibitors via docking screening and molecular dynamics simulation. J. Mol. Graph. Model. 2016, 68, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Park, H.-J. Computer-Aided Discovery of Massonianoside B as a Novel Selective DOT1L Inhibitor. ACS Chem. Biol. 2019, 5, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Somervaille, T.C.P.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef]
- Borkin, D.; Pollock, J.; Kempinska, K.; Purohit, T.; Li, X.; Wen, B.; Zhao, T.; Miao, H.; Shukla, S.; He, M.; et al. Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein-Protein Interaction between Menin and Mixed Lineage Leukemia (MLL). J. Med. Chem. 2016, 59, 892–913. [Google Scholar] [CrossRef]
- Burrows, F.; Wu, T.; Kessler, L.; Li, S.; Zhang, J.; Zarrinkar, P.; Li, L.; Cierpicki, T.; Grembecka, J.; Ren, P.; et al. Abstract LB-A27: A novel small molecule menin-MLL inhibitor for potential treatment of MLL-rearranged leukemias and NPM1/DNMT3A-mutant AML. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef]
- He, S.; Senter, T.J.; Pollock, J.; Han, C.; Upadhyay, S.K.; Purohit, T.; Gogliotti, R.D.; Lindsley, C.W.; Cierpicki, T.; Stauffer, S.R.; et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J. Med. Chem. 2014, 57, 1543–1556. [Google Scholar] [CrossRef]
- Senter, T.; Gogliotti, R.D.; Han, C.; Locuson, C.W.; Morrison, R.; Daniels, J.S.; Cierpicki, T.; Grembecka, J.; Lindsley, C.W.; Stauffer, S.R. Progress towards small molecule menin-mixed lineage leukemia (MLL) interaction inhibitors with in vivo utility. Bioorg. Med. Chem. Lett. 2015, 25, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Aguilar, A.; Xu, T.; Zheng, K.; Huang, L.; Stuckey, J.; Chinnaswamy, K.; Bernard, D.; Fernández-Salas, E.; Liu, L.; et al. Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein–Protein Interaction. Angew. Chem. Int. Ed. 2018, 57, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Dafflon, C.; Craig, V.J.; Méreau, H.; Gräsel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Lam, E.Y.N.; Becher, I.; Lugo, D.; Cannizzaro, E.; Joberty, G.; Ward, A.; Wiese, M.; Fong, C.Y.; Ftouni, S.; et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 2016, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Futur. Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef]
- Kharenko, O.A.; Hansen, H.C. Novel approaches to targeting BRD4. Drug Discov. Today Technol. 2017, 24, 19–24. [Google Scholar] [CrossRef]
- Huang, W.; Zheng, X.; Yang, Y.; Wang, X.; Shen, Z. An Overview on Small Molecule Inhibitors of BRD4. Mini Rev. Med. Chem. 2016, 16, 1403–1414. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef]
- Pericole, F.V.; Lazarini, M.; de Paiva, L.B.; da Duarte, A.S.S.; Vieira Ferro, K.P.; Niemann, F.S.; Roversi, F.M.; Olalla Saad, S.T. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Yang, Y.; Yang, L.; Tian, J.; Zhang, F.; Zhou, J.; Zhang, H. 3-Hydroxyisoindolin-1-one derivates: Synthesis by palladium-catalyzed CH activation as BRD4 inhibitors against human acute myeloid leukemia (AML) cells. Bioorg. Chem. 2019, 86, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Tontsch-Grunt, U.; Rudolph, D.; Waizenegger, I.; Baum, A.; Gerlach, D.; Engelhardt, H.; Wurm, M.; Savarese, F.; Schweifer, N.; Kraut, N. Synergistic activity of BET inhibitor BI 894999 with PLK inhibitor volasertib in AML in vitro and in vivo. Cancer Lett. 2018, 421, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Gudgin, E.J.; Horton, S.J.; Giotopoulos, G.; Meduri, E.; Robson, S.; Cannizzaro, E.; Osaki, H.; Wiese, M.; Putwain, S.; et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 2014, 28, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Townsend, E.C.; Bernard, D.; Dou, Y.; Wang, S. Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J. Med. Chem. 2010, 53, 5179–5185. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-affinity, small-molecule peptidomimetic inhibitors of mll1/wdr5 protein-protein interaction. J. Am. Chem. Soc. 2013, 135, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y. Targeting MLL1 H3K4 Methyltransferase Activity. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef]
- Karatas, H.; Li, Y.; Liu, L.; Ji, J.; Lee, S.; Chen, Y.; Yang, J.; Huang, L.; Bernard, D.; Xu, J.; et al. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J. Med. Chem. 2017, 60, 4818–4839. [Google Scholar] [CrossRef]
- Senisterra, G.; Wu, H.; Allali-Hassani, A.; Wasney, G.A.; Barsyte-Lovejoy, D.; Dombrovski, L.; Dong, A.; Nguyen, K.T.; Smil, D.; Bolshan, Y.; et al. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2012, 449, 151–159. [Google Scholar] [CrossRef]
- Bolshan, Y.; Getlik, M.; Kuznetsova, E.; Wasney, G.A.; Hajian, T.; Poda, G.; Nguyen, K.T.; Wu, H.; Dombrovski, L.; Dong, A.; et al. Synthesis, optimization, and evaluation of novel small molecules as antagonists of WDR5-MLL interaction. ACS Med. Chem. Lett. 2013, 4, 353–357. [Google Scholar] [CrossRef]
- Li, D.D.; Chen, W.L.; Xu, X.L.; Jiang, F.; Wang, L.; Xie, Y.Y.; Zhang, X.J.; Guo, X.K.; You, Q.D.; Sun, H.P. Structure-based design and synthesis of small molecular inhibitors disturbing the interaction of MLL1-WDR5. Eur. J. Med. Chem. 2016, 118, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D.; et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Méreau, H.; De Rijck, J.; Čermáková, K.; Kutz, A.; Juge, S.; Demeulemeester, J.; Gijsbers, R.; Christ, F.; Debyser, Z.; Schwaller, J. Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75). Leukemia 2013, 27, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.D.; Chen, W.L.; Wang, Z.H.; Xie, Y.Y.; Xu, X.L.; Jiang, Z.Y.; Zhang, X.J.; You, Q.D.; Guo, X.K. High-affinity small molecular blockers of mixed lineage leukemia 1 (MLL1)-WDR5 interaction inhibit MLL1 complex H3K4 methyltransferase activity. Eur. J. Med. Chem. 2016, 124, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Tang, L.H.; Shibata, S.; Kelsen, D.P.; Segal, M.; Cheng, C.; Schwartz, G.K.; Doyle, L.A.; Shah, M.A. A multicenter random assignment phase II study of irinotecan and flavopiridol versus irinotecan alone for patients with p53 wild-type gastric adenocarcinoma (NCI 8060). J. Clin. Oncol. Conf. 2012, 30. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid leukemia (AML). Leuk. Res. 2018, 72, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Ishida, T.; Ito, A.; Masaki, A.; Kinoshita, S.; Suzuki, S.; Takino, H.; Yoshida, T.; Ri, M.; Kusumoto, S.; et al. Cyclin-dependent kinase 9 is a novel specific molecular target in adult T-cell leukemia/lymphoma. Blood 2017, 130, 1114–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012, 26, 236–243. [Google Scholar] [CrossRef]
- Xie, S.; Jiang, H.; Zhai, X.W.; Wei, F.; Wang, S.D.; Ding, J.; Chen, Y. Antitumor action of CDK inhibitor LS-007 as a single agent and in combination with ABT-199 against human acute leukemia cells. Acta Pharmacol. Sin. 2016, 37, 1481–1489. [Google Scholar] [CrossRef]
- Yin, T.; Lallena, M.J.; Kreklau, E.L.; Fales, K.R.; Carballares, S.; Torrres, R.; Wishart, G.N.; Ajamie, R.T.; Cronier, D.M.; Iversen, P.W.; et al. A Novel CDK9 Inhibitor Shows Potent Antitumor Efficacy in Preclinical Hematologic Tumor Models. Mol. Cancer Ther. 2014, 13, 1442–1456. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, J.; Wu, Y.; Chen, C.; Zou, F.; Wang, A.; Wu, H.; Hu, Z.; Jiang, Z.; Liu, Q.; et al. Discovery of 4-(((4-(5-chloro-2-(((1s,4s)-4-((2-methoxyethyl)amino)cyclohexyl)amino)pyridin-4-yl)thiazol-2-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile (JSH-150) as a novel highly selective and potent CDK9 kinase inhibitor. Eur. J. Med. Chem. 2018, 158, 896–916. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.T.; Harris, W.J.; Somervaille, T.C.P. LSD1 inhibition: A therapeutic strategy in cancer? Expert Opin. Ther. Targets 2012, 16, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, J.; Scheder, A.-M.; Mohr, S.; Schulz-Fincke, J.; Schmitt, M.; Walter, A.; Tosic, M.; Metzger, E.; Bug, G.; Lübbert, M.; et al. LSD1 Inhibition Leads to Differentiation in Hoxa9/Meis1- but Not in MN1-Induced Acute Myeloid Leukemia. Blood 2017, 130, 3935. [Google Scholar]
- Feng, Z.; Yao, Y.; Zhou, C.; Chen, F.; Wu, F.; Wei, L.; Liu, W.; Dong, S.; Redell, M.; Mo, Q.; et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J. Hematol. Oncol. 2016, 9, 24. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 255. [Google Scholar] [CrossRef] [Green Version]
- Kruger, R.G.; Mohammad, H.; Smitheman, K.; Cusan, M.; Liu, Y.; Pappalardi, M.; Federowicz, K.; Van Aller, G.; Kasparec, J.; Tian, X.; et al. Inhibition of LSD1 as a therapeutic strategy for the treatment of acute myeloid leukemia. Blood 2013, 122, 3964. [Google Scholar]
- Lillico, R.; Lawrence, C.K.; Lakowski, T.M. Selective DOT1L, LSD1, and HDAC Class i Inhibitors Reduce HOXA9 Expression in MLL-AF9 Rearranged Leukemia Cells, but Dysregulate the Expression of Many Histone-Modifying Enzymes. J. Proteome Res. 2018, 17, 2657–2667. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.T.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugino, N.; Kawahara, M.; Tatsumi, G.; Kanai, A.; Matsui, H.; Yamamoto, R.; Nagai, Y.; Fujii, S.; Shimazu, Y.; Hishizawa, M.; et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 2017, 31, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, W.; Li, S.; Li, S.; Liu, J.; Xing, Y.; Zhang, S.; Zhou, J.Z.; Xing, H.; Xu, Y.; et al. Identification of JL1037 as a novel, specific, reversible lysine-specific demethylase 1 inhibitor that induce apoptosis and autophagy of AML cells. Oncotarget 2017, 8, 31901–31914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, D.P.; Alli, C.; Bremberg, U.; Cartic, S.; Jordan, A.M.; Geitmann, M.; Maiques-Diaz, A.; McGonagle, A.E.; Somervaille, T.C.P.; Spencer, G.J.; et al. Development of (4-Cyanophenyl)glycine Derivatives as Reversible Inhibitors of Lysine Specific Demethylase 1. J. Med. Chem. 2017, 60, 7984–7999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.B.C.; Bachovchin, D.A.; Brown, S.J.; Finn, M.G.; Rosen, H.; Cravatt, B.F.; Mowen, K.A. Novel inhibitors for PRMT1 discovered by high-throughput screening using activity-based fluorescence polarization. ACS Chem. Biol. 2012, 7, 1198–1204. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, H.; Jin, J.; Xie, Y.; Chen, Z.; Zhang, H.; Lian, F.; Liu, Y.C.; Zhang, C.; Ding, H.; et al. Development of Potent Type i Protein Arginine Methyltransferase (PRMT) Inhibitors of Leukemia Cell Proliferation. J. Med. Chem. 2017, 60, 8888–8905. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, D.G.; Xu, H.; Chen, C.W.; Hoshii, T.; Eisold, M.E.; Delaney, C.; Cusan, M.; Deshpande, A.J.; Huang, C.H.; Lujambio, A.; et al. Histone acetyltransferase activity of MOF is required for MLL-AF9 leukemogenesis. Cancer Res. 2017, 77, 1753–1762. [Google Scholar] [CrossRef]
- Munier-Lehmann, H.; Vidalain, P.O.; Tangy, F.; Janin, Y.L. On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 2013, 56, 3148–3167. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers 2019, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Rathod, P.K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord. Drug Targets 2010, 10, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Leukemia, M.; Sykes, D.B.; Kfoury, Y.S.; Mercier, E.; Janzer, A.; Schreiber, S.L.; Scadden, D.T. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 2016, 167, 171–186. [Google Scholar] [CrossRef]
- Schnittger, S.; Haferlach, C.; Ulke, M.; Alpermann, T.; Kern, W.; Haferlach, T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 2010, 116, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; de Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O.; et al. Leucémies Aiguës Myéloblastiques de l’Enfant (LAME) Cooperative Groups Incidence and prognostic impact of c-Kit, FLT3 and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006, 20, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Patel, K.P.; Garcia-Manero, G.; Luthra, R.; Pierce, S.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pemmaraju, N.; Konopleva, M.; et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk. Lymphoma 2014, 55, 1925–1929. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Heyn, H.; Vizoso, M.; Moutinho, C.; Vidal, E.; Gomez, A.; Martínez-Cardús, A.; Simó-Riudalbas, L.; Moran, S.; Jost, E.; et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene 2016, 35, 3079–3082. [Google Scholar] [CrossRef]
- Oh, S.; Lee, M.; Lee, J.; Kim, H.; Lee, J.; Lee, S.; Park, T.; Lee, J. Mutational study of DNMT3A in acute myeloid leukemia. Haematologica 2013, 98, 398. [Google Scholar]
- Sandoval, J.E.; Huang, Y.-H.; Muise, A.; Goodell, M.A.; Reich, N.O. Mutations in the DNMT3A DNA methyltransferase in AML patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Yang, N.; Sharma, K.K.; Balys, M.; Corbett, C.A.; Jordan, C.T.; Becker, M.W.; Steidl, U.; Abdel-Wahab, O.; Levine, R.L.; et al. Selective Activity of the Histone Deacetylase Inhibitor AR-42 against Leukemia Stem Cells: A Novel Potential Strategy in Acute Myelogenous Leukemia. Mol. Cancer Ther. 2014, 13, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojo, I.; Jiemjit, A.; Trepel, J.B.; Sparreboom, A.; Figg, W.D.; Rollins, S.; Tidwell, M.L.; Greer, J.; Eun, J.C.; Lee, M.J.; et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2007, 109, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Gjertsen, B.T.; Bruserud, Ø. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenet. 2013, 5, 12. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morabito, F.; Voso, M.T.; Hohaus, S.; Gentile, M.; Vigna, E.; Recchia, A.G.; Iovino, L.; Benedetti, E.; Lo-Coco, F.; Galimberti, S. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin. Investig. Drugs 2016, 25, 1117–1131. [Google Scholar] [CrossRef]
- Kirschbaum, M.H.; Foon, K.A.; Frankel, P.; Ruel, C.; Pulone, B.; Tuscano, J.M.; Newman, E.M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A California Cancer Consortium Study. Leuk. Lymphoma 2014, 55, 2301–2304. [Google Scholar] [CrossRef]
- Di Matteo, A.; Franceschini, M.; Chiarella, S.; Rocchio, S.; Travaglini-Allocatelli, C.; Federici, L. Molecules that target nucleophosmin for cancer treatment: An update. Oncotarget 2016, 7, 44821–44840. [Google Scholar] [CrossRef]
- Yi, S.; Wen, L.; He, J.; Wang, Y.; Zhao, F.; Zhao, J.; Zhao, Z.; Cui, G.; Chen, Y. Deguelin, a selective silencer of the NPM1 mutant, potentiates apoptosis and induces differentiation in AML cells carrying the NPM1 mutation. Ann. Hematol. 2015, 94, 201–210. [Google Scholar] [CrossRef]
- Wang, Y.; Yi, S.; Wen, L.; Zhang, B.; Zhao, Z.; Hu, J.; Zhao, F.; He, J.; Fang, J.; Zhang, C.; et al. Nontoxic-dose of Deguelin Induce NPMc+ AML Cell Differentiation by Selectively Targeting Mt NPM1/SIRT1 Instead of HDAC1/3. Curr. Cancer Drug Targets 2014, 14, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, H.L.; Cui, G.H.; Wu, Q.L.; He, J.; Chen, W.H. [Deguelin regulates cell cycle and nuclear pore complex protein Nup98 and Nup88 in U937 cells in vitro]. Zhonghua Xue Ye Xue Za Zhi 2007, 28, 115–118. [Google Scholar] [PubMed]
- Qi, W.; Shakalya, K.; Stejskal, A.; Goldman, A.; Beeck, S.; Cooke, L.; Mahadevan, D. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene 2008, 27, 4210–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balusu, R.; Fiskus, W.; Rao, R.; Chong, D.G.; Nalluri, S.; Mudunuru, U.; Ma, H.; Chen, L.; Venkannagari, S.; Ha, K.; et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood 2011, 118, 3096–3106. [Google Scholar] [CrossRef] [PubMed]
- Nabbouh, A.I.; Hleihel, R.S.; Saliba, J.L.; Karam, M.M.; Hamie, M.H.; Wu, H.C.J.M.; Berthier, C.P.; Tawil, N.M.; Bonnet, P.A.A.; Deleuze-Masquefa, C.; et al. Imidazoquinoxaline derivative EAPB0503: A promising drug targeting mutant nucleophosmin 1 in acute myeloid leukemia. Cancer 2017, 123, 1662–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRonde-LeBlanc, N.A.; Wolberger, C. Structure of HoxA9 and Pbx1 bound to DNA: Hox hexapeptide and DNA recognition anterior to posterior. Genes Dev. 2003, 17, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Knoepfler, P.S.; Lu, Q.; Kamps, M.P. Pbx1-Hox heterodimers bind DNA on inseparable half-sites that permit intrinsic DNA binding specificity of the Hox partner at nucleotides 3′ to a TAAT motif. Nucleic Acids Res. 1996, 24, 2288–2294. [Google Scholar] [CrossRef]
- Neuteboom, S.T.; Murre, C. Pbx raises the DNA binding specificity but not the selectivity of antennapedia Hox proteins. Mol. Cell. Biol. 2015, 17, 4696–4706. [Google Scholar] [CrossRef]
- Chang, C.P.; Shen, W.F.; Rozenfeld, S.; Lawrence, H.J.; Largman, C.; Cleary, M.L. Pbx proteins display hexapeptide-dependent cooperative DNA binding with a subset of Hox proteins. Genes Dev. 1995, 9, 663–674. [Google Scholar] [CrossRef]
- Neuteboom, S.T.; Peltenburg, L.T.; van Dijk, M.A.; Murre, C. The hexapeptide LFPWMR in Hoxb-8 is required for cooperative DNA binding with Pbx1 and Pbx2 proteins. Proc. Natl. Acad. Sci. USA 2006, 92, 9166–9170. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Sykes, D.B.; Gay, G.L.; Sykes, B.D. The HoxB1 hexapeptide is a prefolded domain: Implications for the Pbx1/Hox interaction. Protein Sci. 2002, 10, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Sprules, T.; Green, N.; Featherstone, M.; Gehring, K. Conformational changes in the PBX homeodomain and C-terminal extension upon binding DNA and HOX-derived YPWM peptides. Biochemistry 2000, 39, 9943–9950. [Google Scholar] [CrossRef] [PubMed]
- Zeiske, T.; Baburajendran, N.; Kaczynska, A.; Shapiro, L.; Honig, B.; Mann, R.S. Intrinsic DNA Shape Accounts for Affinity Differences between Hox-Cofactor Binding Sites Correspondence Data and Software Availability 5ZJQ. Cell Rep. 2018, 24, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Jennings, P.A.; Murre, C. Conformational changes induced in Hoxb-8/Pbx-1 heterodimers in solution and upon interaction with specific DNA. Mol. Cell. Biol. 2015, 17, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Dard, A.; Jia, Y.; Reboulet, J.; Bleicher, F.; Lavau, C.; Merabet, S. The human HOXA9 protein uses paralog-specific residues of the homeodomain to interact with TALE-class cofactors. Sci Rep. 2019, 9, 5664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.; Pirard, P.M.; Shears, L.; Sohal, J.; Pettengell, R.; Pandha, H.S. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res. 2007, 67, 5806–5813. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Neacato, I.I.; Rodríguez, J.A.; Pandha, H.S.; Morgan, R.; Penichet, M.L. Disruption of HOX activity leads to cell death that can be enhanced by the interference of iron uptake in malignant B cells. Leukemia 2010, 24, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.; El-Tanani, M.; Hunter, K.D.; Harrington, K.J.; Pandha, H.S. Targeting HOX/PBX dimers in cancer. Oncotarget 2017, 8, 32322–32331. [Google Scholar] [CrossRef] [Green Version]
- Errico, M.C.; Felicetti, F.; Bottero, L.; Mattia, G.; Boe, A.; Felli, N.; Petrini, M.; Bellenghi, M.; Pandha, H.S.; Calvaruso, M.; et al. The abrogation of the HOXB7/PBX2 complex induces apoptosis in melanoma through the miR-221&222-c-FOS pathway. Int. J. Cancer 2013, 133, 879–892. [Google Scholar]
- Shears, L.; Plowright, L.; Harrington, K.; Pandha, H.S.; Morgan, R. Disrupting the Interaction Between HOX and PBX Causes Necrotic and Apoptotic Cell Death in the Renal Cancer Lines CaKi-2 and 769-P. J. Urol. 2008, 180, 2196–2201. [Google Scholar] [CrossRef]
- Morgan, R.; Plowright, L.; Harrington, K.J.; Michael, A.; Pandha, H.S. Targeting HOX and PBX transcription factors in ovarian cancer. BMC Cancer 2010, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Kelly, Z.; Moller-Levet, C.; McGrath, S.; Butler-Manuel, S.; Kavitha Madhuri, T.; Kierzek, A.M.; Pandha, H.; Morgan, R.; Michael, A. The prognostic significance of specific HOX gene expression patterns in ovarian cancer. Int. J. Cancer 2016, 139, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Boxall, A.; Harrington, K.J.; Simpson, G.R.; Gillett, C.; Michael, A.; Pandha, H.S. Targeting the HOX/PBX dimer in breast cancer. Breast Cancer Res. Treat. 2012, 136, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.; Boxall, A.; Harrington, K.J.; Simpson, G.R.; Michael, A.; Pandha, H.S. Targeting HOX transcription factors in prostate cancer. BMC Urol. 2014, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Simpson, G.; Gray, S.; Gillett, C.; Tabi, Z.; Spicer, J.; Harrington, K.J.; Pandha, H.S. HOX transcription factors are potential targets and markers in malignant mesothelioma. BMC Cancer 2016, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- Platais, C.; Radhakrishnan, R.; Ebensberger, S.N.; Morgan, R.; Lambert, D.W.; Hunter, K.D. Targeting HOX-PBX interactions causes death in oral potentially malignant and squamous carcinoma cells but not normal oral keratinocytes. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-Y.; Zhou, T.; Du, Y.-B.; Shi, Q.; Chen, K.-N. Targeting HOX/PBX dimer formation as a potential therapeutic option in Esophageal Squamous Cell Carcinoma. Cancer Sci. 2019. [Google Scholar] [CrossRef]
- Li, Z.; Huang, H.; Li, Y.; Jiang, X.; Chen, P.; Arnovitz, S.; Radmacher, M.D.; Maharry, K.; Elkahloun, A.; Yang, X.; et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood 2012, 119, 2314–2324. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, Z.; Li, Y.; Arnovitz, S.; Chen, P.; Huang, H.; Jiang, X.; Hong, G.M.; Kunjamma, R.B.; Ren, H.; et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood 2013, 121, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Primon, M.; Hoffman, E.; Pandha, H.S.; Morgan, R. HTL001, a novel inhibitor of HOX/PBX binding, is highly cytotoxic to prostate and breast cancer cells. Eur. J. Cancer 2016, 69, S133. [Google Scholar] [CrossRef]
- Shi, X.; Bai, S.; Li, L.; Cao, X. Hoxa-9 represses transforming growth factor-β-induced osteopontin gene transcription. J. Biol. Chem. 2001, 276, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Williams, M.E.; Heaton, J.H.; Gelehrter, T.D.; Innis, J.W. Group 13 HOX proteins interact with the MH2 domain of R-Smads and modulate Smad transcriptional activation functions independent of HOX DNA-binding capability. Nucleic Acids Res. 2005, 33, 4475–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Kim, H.G.; Cotta, C.V.; Wan, M.; Tang, Y.; Klug, C.A.; Cao, X. TGFβ/BMP inhibits the bone marrow transformation capability of Hoxa9 by repressing its DNA-binding ability. EMBO J. 2006, 25, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Quéré, R.; Karlsson, G.; Hertwig, F.; Rissler, M.; Lindqvist, B.; Fioretos, T.; Vandenberghe, P.; Slovak, M.L.; Cammenga, J.; Karlsson, S. Smad4 binds Hoxa9 in the cytoplasm and protects primitive hematopoietic cells against nuclear activation by Hoxa9 and leukemia transformation. Blood 2011, 117, 5918–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Thakur, A.R. Two models of smad4 and hoxa9 complex are proposed: Structural and interactional perspective. J. Biomol. Struct. Dyn. 2011, 28, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Pabst, C.; Su, L.; Miller, M.; Liu, F.; Yi, L.; Zhang, R.; Krosl, J.; Yung, E.; Kirschner, J.; et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014, 28, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappano, W.N.; Guo, J.; He, Y.; Ferguson, D.; Jagadeeswaran, S.; Osterling, D.J.; Gao, W.; Spence, J.K.; Pliushchev, M.; Sweis, R.F.; et al. The Histone methyltransferase inhibitor A-366 uncovers a role for G9a/GLP in the epigenetics of leukemia. PLoS ONE 2015, 10, e0131716. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Lambert, M.; David-Cordonnier, M.-H. Targeting Transcription Factor Binding to DNA by Competing with DNA Binders as an Approach for Controlling Gene Expression. Curr. Top. Med. Chem. 2015, 15, 1323–1358. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.Y.; Zhang, J.T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2015, 5. [Google Scholar] [CrossRef]
- Pop, M.S.; Stransky, N.; Garvie, C.W.; Theurillat, J.-P.; Hartman, E.C.; Lewis, T.A.; Zhong, C.; Culyba, E.K.; Lin, F.; Daniels, D.S.; et al. A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein. Mol. Cancer Ther. 2014, 13, 1492–1502. [Google Scholar] [CrossRef]
- Trimmer, E.E.; Zamble, D.B.; Lippard, S.J.; Essigmann, J.M. Human testis-determining factor SRY binds to the major DNA adduct of cisplatin and a putative target sequence with comparable affinities. Biochemistry 1998, 37, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Chválová, K.; Sari, M.A.; Bombard, S.; Kozelka, J. LEF-1 recognition of platinated GG sequences within double-stranded DNA. Influence of flanking bases. J. Inorg. Biochem. 2008, 102, 242–250. [Google Scholar] [CrossRef]
- Chiang, S.Y.; Welch, J.; Beerman, T.A.; Rauscher, F.J. Effects of Minor Groove Binding Drugs on the Interaction of TATA Box Binding Protein and TFIIA with DNA. Biochemistry 1994, 33, 7033–7040. [Google Scholar] [CrossRef]
- Henderson, D.; Hurley, L.H. Specific targeting of protein-DNA complexes by DNA-reactive drugs (+)- CC-1065 and pluramycins. J. Mol. Recognit. 1996, 9, 75–87. [Google Scholar] [CrossRef]
- Minuzzo, M.; Marchini, S.; Broggini, M.; Faircloth, G.; D’Incalci, M.; Mantovani, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc. Natl. Acad. Sci. USA 2000, 97, 6780–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Bible, R.H.; Kottke, T.J.; Svingen, P.A.; Xu, K.; Pang, Y.P.; Hajdu, E.; Kaufmann, S.H. Flavopiridol binds to duplex DNA. Cancer Res. 2000, 60, 2419–2428. [Google Scholar] [PubMed]
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS–FLI1 fusion transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef]
- Osgood, C.L.; Maloney, N.; Kidd, C.G.; Kitchen-Goosen, S.; Segars, L.; Gebregiorgis, M.; Woldemichael, G.M.; He, M.; Sankar, S.; Lessnick, S.L.; et al. Identification of mithramycin analogues with improved targeting of the EWS-FLI1 transcription factor. Clin. Cancer Res. 2016, 22, 4105–4118. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.-R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, H.; Meng, L.; Yin, L. Mithramycin inhibits epithelial-tomesenchymal transition and invasion by downregulating SP1 and SNAI1 in salivary adenoid cystic carcinoma. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Liu, R.; Zhi, X.; Zhou, Z.; Zhang, H.; Yang, R.; Zou, T.; Chen, C. Mithramycin A suppresses basal triple-negative breast cancer cell survival partially via down-regulating Krüppel-like factor 5 transcription by Sp1. Sci. Rep. 2018, 8, 1138. [Google Scholar] [CrossRef]
- Malek, A.; Núñez, L.-E.; Magistri, M.; Brambilla, L.; Jovic, S.; Carbone, G.M.; Morís, F.; Catapano, C.V. Modulation of the Activity of Sp Transcription Factors by Mithramycin Analogues as a New Strategy for Treatment of Metastatic Prostate Cancer. PLoS ONE 2012, 7, e35130. [Google Scholar] [CrossRef]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Erratum: Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor (Cancer Cell (2014) 26(6) (909–922) (S1535610814004231) (10.1016/j.ccell.2014.10.019)). Cancer Cell 2015, 27, 149. [Google Scholar] [CrossRef]
- Zhang, J.P.; Zhang, H.; Wang, H.B.; Li, Y.X.; Liu, G.H.; Xing, S.; Li, M.Z.; Zeng, M.S. Down-regulation of Sp1 suppresses cell proliferation, clonogenicity and the expressions of stem cell markers in nasopharyngeal carcinoma. J. Transl. Med. 2014, 12. [Google Scholar] [CrossRef]
- Yang, F.; Nickols, N.G.; Li, B.C.; Marinov, G.K.; Said, J.W.; Dervan, P.B. Antitumor activity of a pyrrole-imidazole polyamide. Proc. Natl. Acad. Sci. USA 2013, 110, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.F.; Fujiwara, T.; Saito, I.; Sugiyama, H. Sequence-specific alkylation of DNA by duocarmycin A and its novel derivatives bearing Py/Im polyamides. Nucleosides Nucleotides 1999, 18, 1615–1616. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.B.; Doss, R.M.; Marques, M. a Programmable DNA binding oligomers for control of transcription. Curr. Med. Chem. Anticancer Agents 2005, 5, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Donato, L.J.; Brezinski, M.L.; Stafford, R.L.; Hoff, H.; Thorson, J.S.; Dervan, P.B.; Ansari, A.Z. Targeted chemical wedges reveal the role of allosteric DNA modulation in protein—DNA assembly. ACS Chem. Biol. 2008, 3, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Arndt, H.D.; Hauschild, K.E.; Sullivan, D.P.; Lake, K.; Dervan, P.B.; Ansari, A.Z. Toward Artificial Developmental Regulators. J. Am. Chem. Soc. 2003, 125, 13322–13323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.Y.; Tang, Y.; Knosp, W.M.; Stadler, H.S.; Shaw, J.T. Synthesis of diverse lactam carboxamides leading to the discovery of a new transcription-factor inhibitor. Angew. Chem. Int. Ed. 2007, 46, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Depauw, S.; Lambert, M.; Jambon, S.; Paul, A.; Peixoto, P.; Nhili, R.; Marongiu, L.; Figeac, M.; Dassi, C.; Paul-Constant, C.; et al. Heterocyclic Diamidine DNA ligands as HOXA9 Transcription Factor Inhibitors: Design, Molecular Evaluation and Cellular Consequences in HOXA9-Dependant Leukemia Cell Model. J. Med. Chem. 2019, 62, 1306–1329. [Google Scholar] [CrossRef]
- Khutornenko, A.A.; Dalina, A.A.; Chernyak, B.V.; Chumakov, P.M.; Evstafieva, A.G. The Role of Dihydroorotate Dehydrogenase in Apoptosis Induction in Response to Inhibition of the Mitochondrial Respiratory Chain Complex III. Acta Nat. 2014, 6, 69–75. [Google Scholar]
- Christian, S.; Merz, C.; Evans, L.; Gradl, S.; Seidel, H.; Friberg, A.; Eheim, A.; Lejeune, P.; Brzezinka, K.; Zimmermann, K.; et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 2019. [Google Scholar] [CrossRef]
- Madak, J.T.; Bankhead, A.; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef]
- Lewis, T.A.; Sykes, D.B.; Law, J.M.; Muñoz, B.; Rustiguel, J.K.; Nonato, M.C.; Scadden, D.T.; Schreiber, S.L. Development of ML390: A Human DHODH Inhibitor That Induces Differentiation in Acute Myeloid Leukemia. ACS Med. Chem. Lett. 2016, 7, 1112–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaveland, E.S.; Zaharevitz, D.W.; Kelley, J.A.; Paull, K.; Cooney, D.A.; Ford, H. Identification of a novel inhibitor (NSC 665564) of dihydroorotate dehydrogenase with a potency equivalent to brequinar. Biochem. Biophys. Res. Commun. 1996, 223, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, W.; Chen, W.; Lian, F.; Lang, L.; Huang, Y.; Xu, Y.; Zhang, N.; Chen, Y.; Liu, M.; et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica 2018, 103, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Weetall, M.; Trotta, C.; Cintron, K.; Ma, J.; Kim, M.J.; Furia, B.; Romfo, C.; Graci, J.D.; Li, W.; et al. Targeting of Hematologic Malignancies with PTC299, A Novel Potent Inhibitor of Dihydroorotate Dehydrogenase with Favorable Pharmaceutical Properties. Mol. Cancer Ther. 2019, 18, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Armstrong, S.A. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 2015, 43, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Choi, M.; Kim, J.-E. The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle. Cell Cycle 2014, 13, 726–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011, 21, 1370–1373. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Kim, R.; Park, G.; Park, J.-W.; Kim, J.-E. Deficiency of H3K79 Histone Methyltransferase Dot1-like Protein (DOT1L) Inhibits Cell Proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Cramer, S.A.; Adjei, I.M.; Labhasetwar, V. Advancements in the delivery of epigenetic drugs. Expert Opin. Drug Deliv. 2015, 12, 1501–1512. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Crook, T. Cancer Epigenetics: New Therapies and New Challenges. J. Drug Deliv. 2013, 2013, 529312. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Stapnes, C.; Ersvær, E.; Gjertsen, B.; Ryningen, A. Histone Deacetylase Inhibitors in Cancer Treatment: A Review of the Clinical Toxicity and the Modulation of Gene Expression in Cancer Cells. Curr. Pharm. Biotechnol. 2007, 8, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Jerković, I.; Ibrahim, D.M.; Andrey, G.; Haas, S.; Hansen, P.; Janetzki, C.; González Navarrete, I.; Robinson, P.N.; Hecht, J.; Mundlos, S. Genome-Wide Binding of Posterior HOXA/D Transcription Factors Reveals Subgrouping and Association with CTCF. PLoS Genet. 2017, 13, e1006567. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhao, G.-N.; Lv, X.; Liu, G.; Wang, L.Y.; Hao, D.-L.; Wang, J.; Liu, D.-P.; Liang, C.-C. CTCF Controls HOXA Cluster Silencing and Mediates PRC2-Repressive Higher-Order Chromatin Structure in NT2/D1 Cells. Mol. Cell. Biol. 2014, 34, 3867–3879. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Sobh, A.; Vulpe, C.D.; Brewer, E.; Dovat, S.; Qiu, Y.; Huang, S. HOX Loci Focused CRISPR/sgRNA Library Screening Identifying Critical CTCF Boundaries. J. Vis. Exp. 2019, 31, 145. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, H.; Chen, P.; He, M.; Li, Y.; Arnovitz, S.; Jiang, X.; He, C.; Hyjek, E.; Zhang, J.; et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat. Commun. 2012, 2, 688. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, A.; Careccia, S.; Lulli, V.; Romania, P.; Marziali, G.; Testa, U.; Lavorgna, S.; Lo-Coco, F.; Petti, M.C.; Calabretta, B.; et al. MiRNA let-7c promotes granulocytic differentiation in acute myeloid leukemia. Oncogene 2013, 32, 3648–3654. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, H.; Li, Z.; He, C.; Li, Y.; Chen, P.; Gurbuxani, S.; Arnovitz, S.; Hong, G.-M.; Price, C.; et al. miR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 19397–19402. [Google Scholar] [CrossRef]
- Luo, H.; Zhu, G.; Zha, J.; Yan, B.; Guo, Y.; Xu, J.; Lai, Q.; Xu, B.; Yang, F.-C.; Li, W.; et al. Activation of Hottip LncRNA Perturbs HSC Function Leading to AML like Disease in Mice. Blood 2018, 132, 3877. [Google Scholar] [CrossRef]
- Xiong, Y.; Li, Z.; Ji, M.; Tan, A.C.; Bemis, J.; Tse, J.V.; Huang, G.; Park, J.; Ji, C.; Chen, J.; et al. MIR29B regulates expression of MLLT11 (AF1Q), an MLL fusion partner, and low MIR29B expression associates with adverse cytogenetics and poor overall survival in AML. Br. J. Haematol. 2011, 153, 753–757. [Google Scholar] [CrossRef]
- Chen, P.; Price, C.; Li, Z.; Li, Y.; Cao, D.; Wiley, A.; He, C.; Gurbuxani, S.; Kunjamma, R.B.; Huang, H.; et al. miR-9 is an essential oncogenic microRNA specifically overexpressed in mixed lineage leukemia-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 11511–11516. [Google Scholar] [CrossRef] [PubMed]
- Primon, M.; Hunter, K.D.; Pandha, H.S.; Morgan, R. Kinase Regulation of HOX Transcription Factors. Cancers 2019, 11, 508. [Google Scholar] [CrossRef] [PubMed]
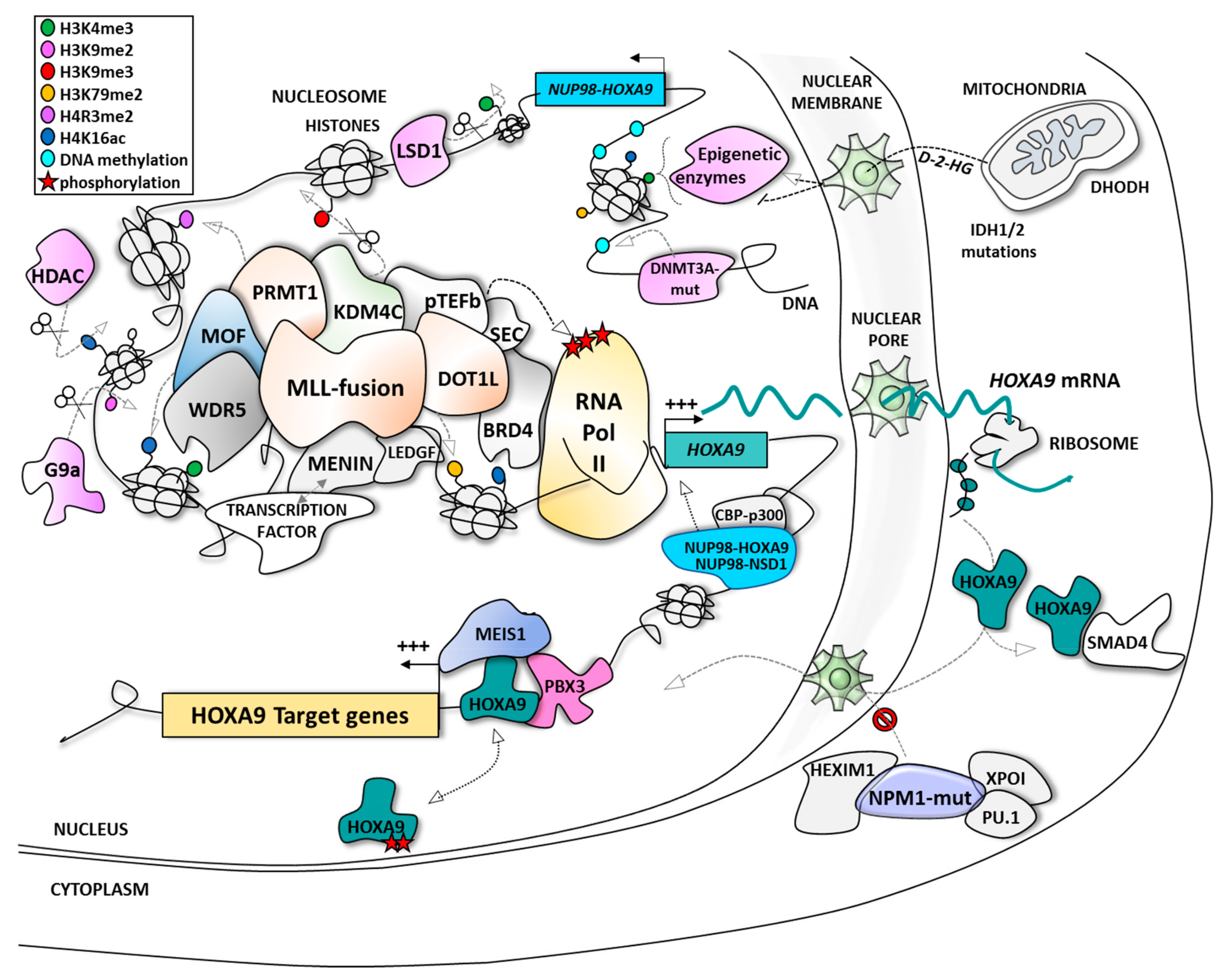
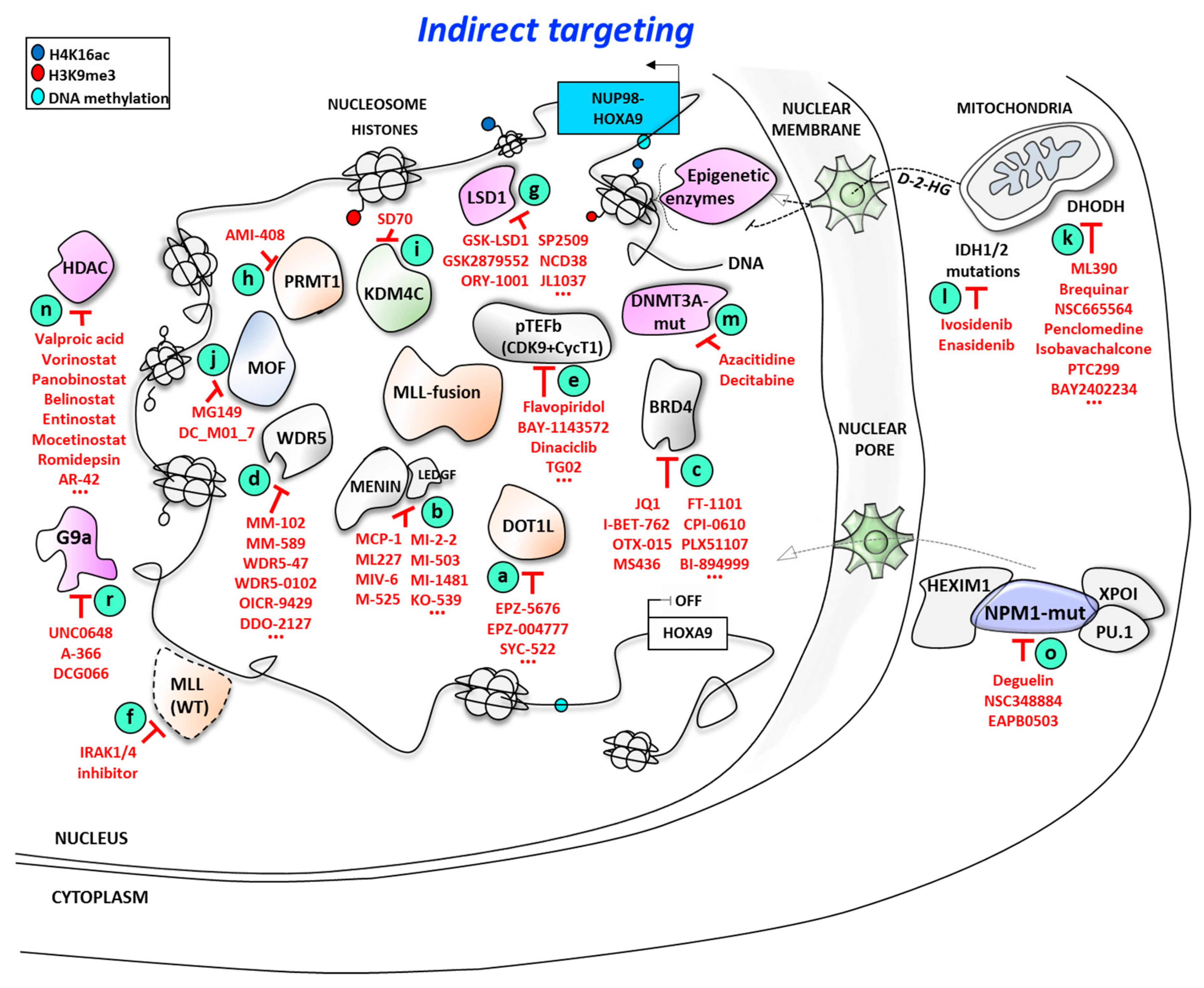
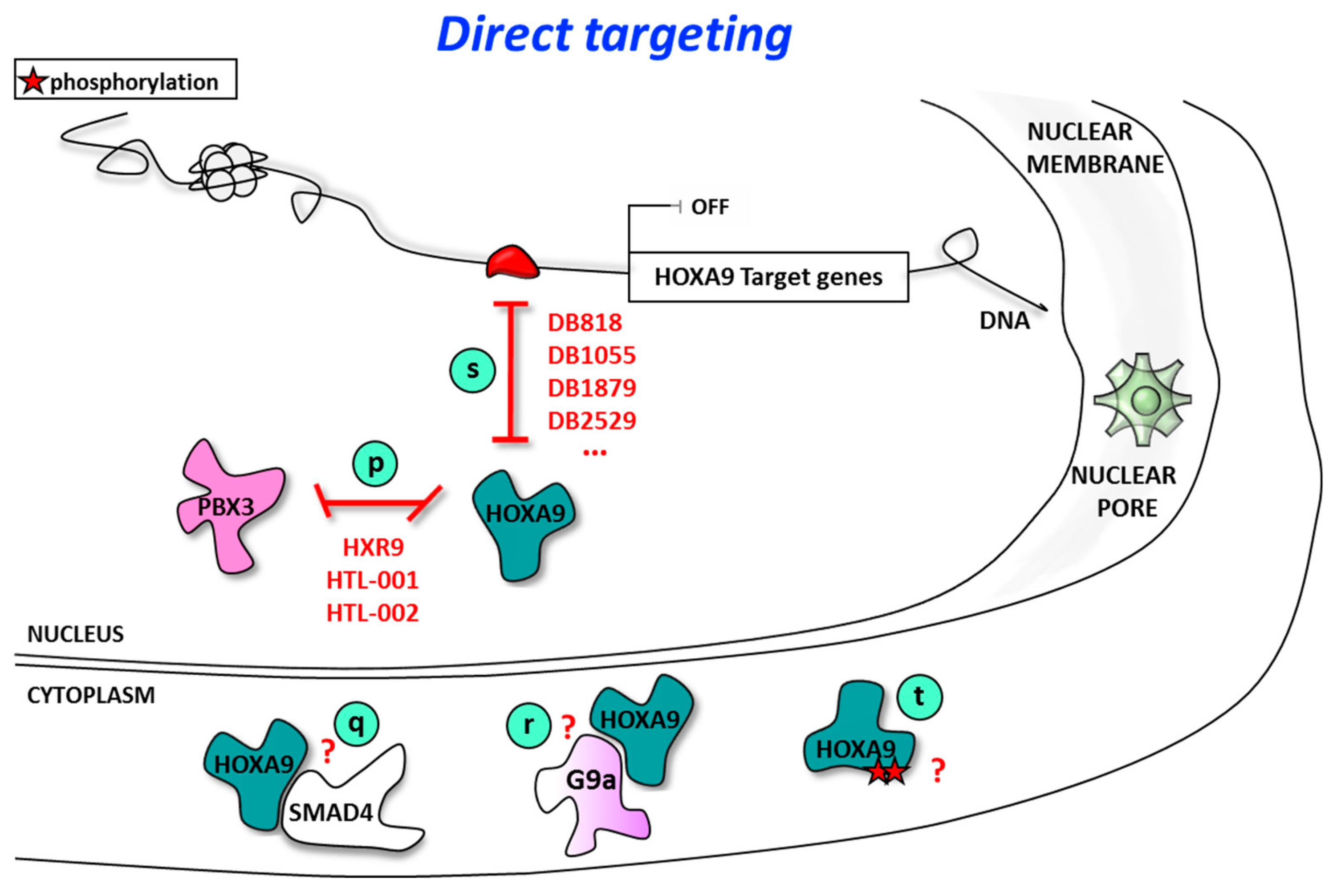

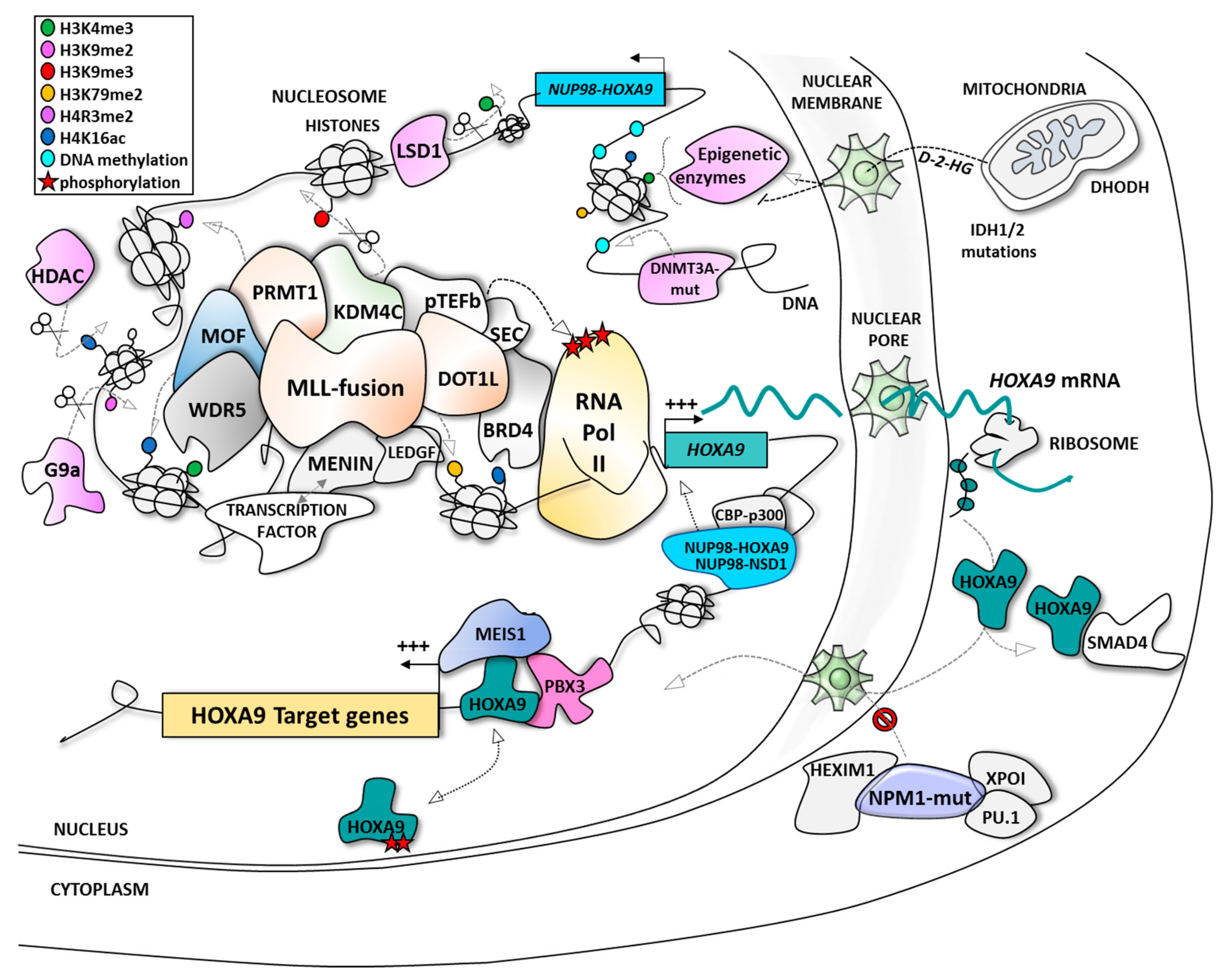{kind=link}
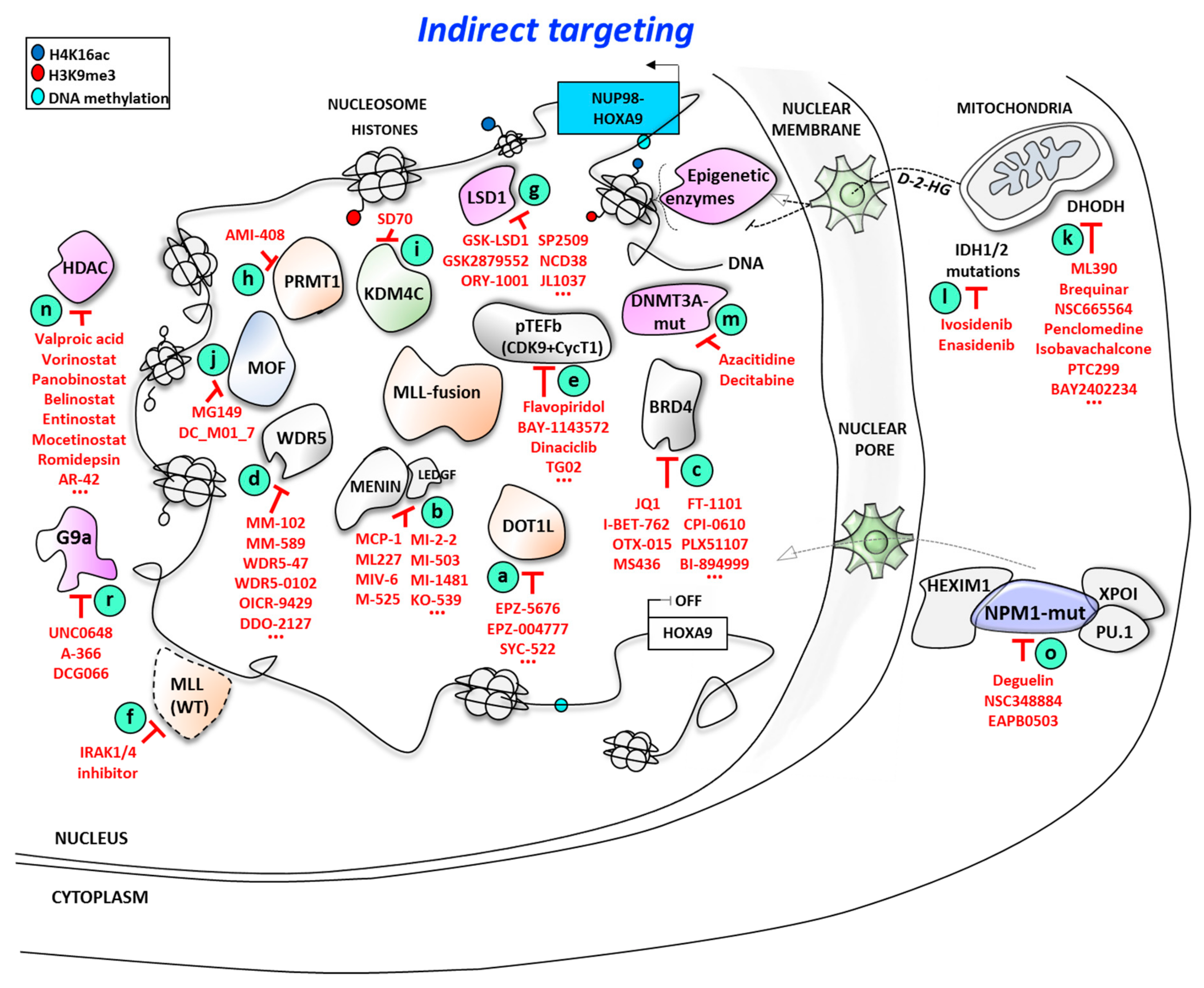{kind=link}
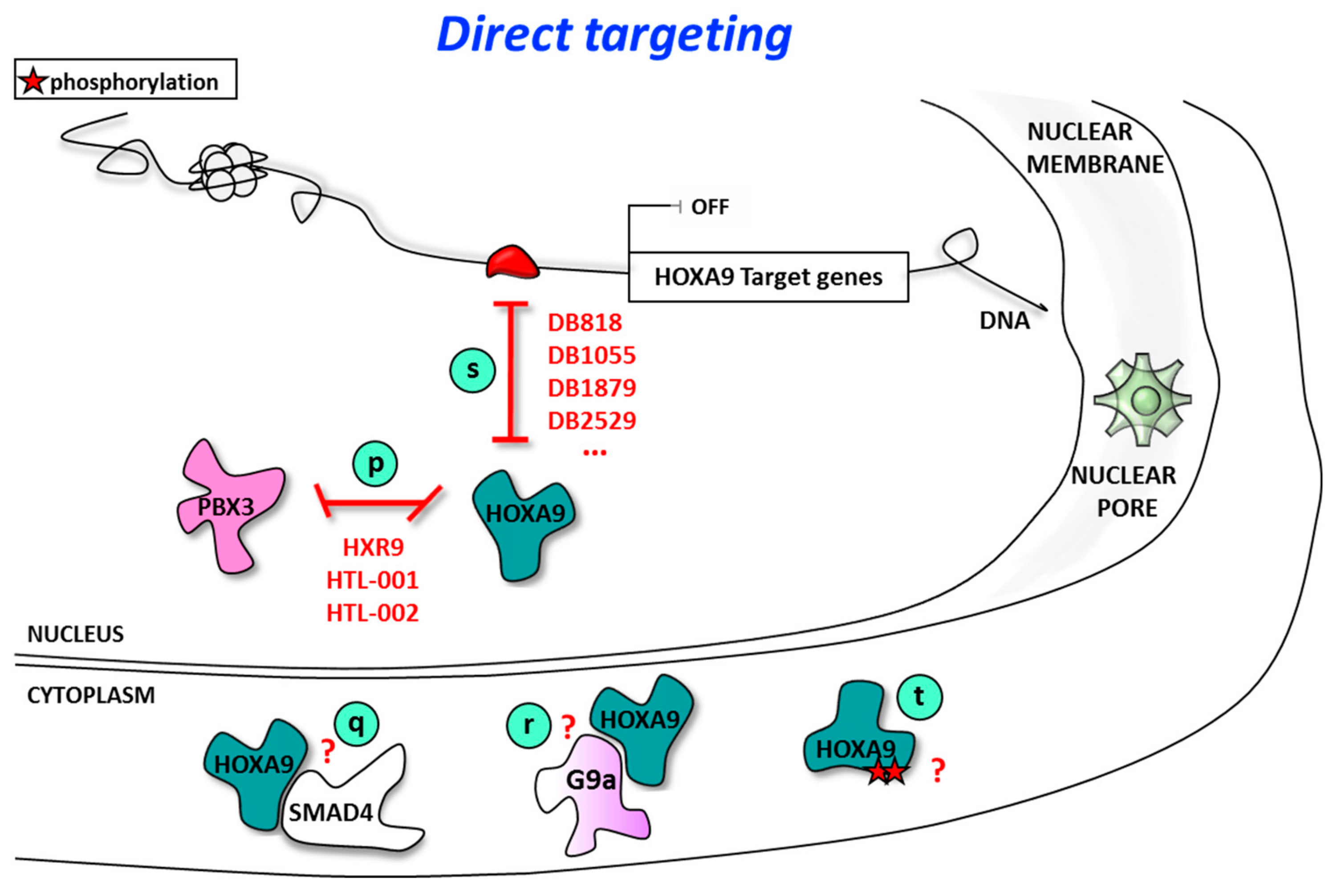{kind=link}
| Type of Alteration | Fusion/Mutation/Additional Chromosome | Translocation/Inversion/Deletion | References |
|---|---|---|---|
| Chromosomal alterations | MLL fusions | 11q23 translocations | [43,45,47] |
| NUP98-NSD1 | t(5;11)(q35;p15) | [80,82] | |
| NUP98-HOXA9 | t(7;11)(p15;p15) | [83] | |
| NUP98-HOXA10 | t(7;11)(p15;p15) | [84] | |
| NUP98-HOXC11 | t(11;12)(p15;q13) | [85] | |
| NUP98-HOXD11 | t(2;11)(q31;p15) | [86] | |
| NUP98-HOXD13 | t(2;11)(q31;p15) | [84] | |
| NUP98-HHEX | t(10;11)(q23;p15) | [87] | |
| NUP98-KDM5A | t(11;12)(p15;p13) | [33] | |
| NUP98-PHF23 | t(11;17)(p15;p13) | [33] | |
| NUP98-PRRX1 | t(1;11)(q24;p15) | [33] | |
| NUP98-DDX10 | inv(11)(p15q22) | [33] | |
| MYST3-CREBBP | t(8;16)(p11;p13) | [88] | |
| RUNX1-EVI1 | t(3;21)(q26;q22) | [89] | |
| CDX2-ETV6 | t(12;13)(p13;q12) | [90] | |
| CALM-AF10 | t(10;11)(p12-14;q14-21) | [91] | |
| SET-NUP214 | del(9)(q34.11;q34.13) | [92] | |
| NPM1-MLF1 | t(3;5)(q25;q34) | [93,94] | |
| +8 | / | [81] | |
| Mutations | NPM1 | [48,49,50,75] | |
| MLL-PTD | [42] | ||
| DNMT3A | [95] | ||
| EZH2 | [42] | ||
| IDH1/2 | [50,96] | ||
| Polymorphism | GFI1-S36N | [97] |
| Type of Targeting | Target | Inhibitor | Stage of development | References | ||
|---|---|---|---|---|---|---|
| Indirect Targeting of HOXA9 | Epigenetic Control of HOXA9 expression | Proteins interaction within the MLL Complex | DOT1L | EPZ-5676 | Phase I | [128,129,130,131,132,133,134] |
| EPZ004777 | Preclinical Stage | [129,130,131,132,133] | ||||
| SYC-522 | [138] | |||||
| Cpd 9e | [139] | |||||
| Cpd 3, 9 | [140] | |||||
| DC_L115 | [141] | |||||
| Cpd 12, 13 | [142] | |||||
| L01, L03, L04, L05 | [143] | |||||
| Massonianoside B | [144] | |||||
| Menin/ LEDGF | KO-539 | Preclinical Stage | [149] | |||
| MCP-1 | [147] | |||||
| MI-2-2 | [146] | |||||
| MI-463 | [147] | |||||
| MI-503 | [147] | |||||
| MI-538 | [148] | |||||
| MI-1481 | [148] | |||||
| ML227 | [151] | |||||
| MIV-6 | [152] | |||||
| M-525 | [152] | |||||
| BRD4 | I-BET762 | Phase I/II | [156,157] | |||
| OTX015 | Phase I/Ib | |||||
| CPI-0610 | Phase I/II | |||||
| FT-1101 | Phase I | |||||
| BI-894999 | Phase I | |||||
| BMS-986158 | Phase I/II (2019) | |||||
| PLX51107 | Phase I | |||||
| Indirect Targeting of HOXA9 | Epigenetic Control of HOXA9 expression | Proteins interaction within the MLL Complex (continued) | BRD4 | RO6870810 | Phase I | [156,157] |
| GSK2820151 | Phase I | |||||
| (+)-JQ | Preclinical Stage | [159,160,161,162,163] | ||||
| MS436 | ||||||
| Ir-based cpd1a | ||||||
| Isoindoline derivate 10e | [161] | |||||
| WDR5 | MM-102 | Preclinical Stage | [166] | |||
| MM-401 | [167] | |||||
| MM-589 | [168] | |||||
| WDR5-0102 | [169] | |||||
| WDR5-47 | [170] | |||||
| Cpd 23 | [171] | |||||
| OICR-9429 | [172] | |||||
| DDO-2117 | [173] | |||||
| CDK9 | Flavopiridol | Phase I/II | [175,176] | |||
| BAY-1143572 | Phase I | [177] | ||||
| Dinaciclib | Phase I/II/III | [178] | ||||
| TG02 | Phase I | [179] | ||||
| LS-007 | Preclinical Stage | [180] | ||||
| LY2857785 | [181] | |||||
| JSH-150 | [182] | |||||
| Other epigenetic modulators | LSD1 | Tranylcypromine (TCP) | Phase I | [187,188] | ||
| GSK2879552 | [187,188] | |||||
| ORY-1001 | [188,190] | |||||
| Cyclopropyl cpds | Preclinical Stage | [186] | ||||
| SP2509 | [191] | |||||
| NCD38 | [192] | |||||
| JL1037 | [193] | |||||
| Cpd 32 | [194] | |||||
| PRMT1 | AMI-408 | Preclinical Stage | [196] | |||
| DCPR049_12 | [197] | |||||
| KDM4C | SD70 | Preclinical Stage | [198] | |||
| MOF | MG149 | Preclinical Stage | [199] | |||
| DNMT3A | Azacitidine | FDA approval in AML | [212] | |||
| Decitabine | ||||||
| HDAC | Valproic acid | Phase I/II in AML | [213,214,215,216,217,218] | |||
| SAHA | ||||||
| LBH589 | ||||||
| Romidepsin | ||||||
| Belinostat | ||||||
| Entinostat | ||||||
| Mocetinostat | ||||||
| AR-42 | ||||||
| G9a | UNC0648 | Preclinical Stage | [256] | |||
| A-366 | [257] | |||||
| Metabolism proteins | DHODH | Brequinar | Phase II | [288] | ||
| BAY2402234 | Phase I | [289] | ||||
| ASLAN003 | Phase II | [290] | ||||
| ML390 | Preclinical Stage | [291] | ||||
| NSC665564 | [292] | |||||
| Isobavachalcone | [293] | |||||
| PTC299 | Preclinical Stage | [294] | ||||
| IDH1 | Ivosidenib | FDA approval in AML | [207,208] | |||
| IDH2 | Enasidenib | FDA approval in AML | [206] | |||
| Other regulators of HOXA9 expression | NPM1c+ | Deguelin | Preclinical Stage | [222] | ||
| NSC348884 | [223,224] | |||||
| EAPB0503 | [225] | |||||
| Degradation | MLL | IRAK4 inhibitor | Preclinical Stage | [122] | ||
| Direct HOXA9 Targeting | Protein/protein interaction | HOX/PBX interaction | HXR9 | Phase I | [70,75,248,249] | |
| HTL-001 | Phase I (end 2019) | [250] | ||||
| HTL-002 | Preclinical Stage | [HOX Therapeutics Ltd.] | ||||
| Protein/ DNA interaction | HOXA9 DNA binding site | DB818 | Preclinical Stage | [287] | ||
| DB1055 | ||||||
| DB1879 | ||||||
| DB2529 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, M.; Alioui, M.; Jambon, S.; Depauw, S.; Seuningen, I.V.; David-Cordonnier, M.-H. Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers 2019, 11, 837. https://doi.org/10.3390/cancers11060837
Lambert M, Alioui M, Jambon S, Depauw S, Seuningen IV, David-Cordonnier M-H. Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers. 2019; 11(6):837. https://doi.org/10.3390/cancers11060837
Chicago/Turabian StyleLambert, Mélanie, Meryem Alioui, Samy Jambon, Sabine Depauw, Isabelle Van Seuningen, and Marie-Hélène David-Cordonnier. 2019. "Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia" Cancers 11, no. 6: 837. https://doi.org/10.3390/cancers11060837
APA StyleLambert, M., Alioui, M., Jambon, S., Depauw, S., Seuningen, I. V., & David-Cordonnier, M.-H. (2019). Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers, 11(6), 837. https://doi.org/10.3390/cancers11060837