Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction of Gastrointestinal Stromal Tumors (GISTs)
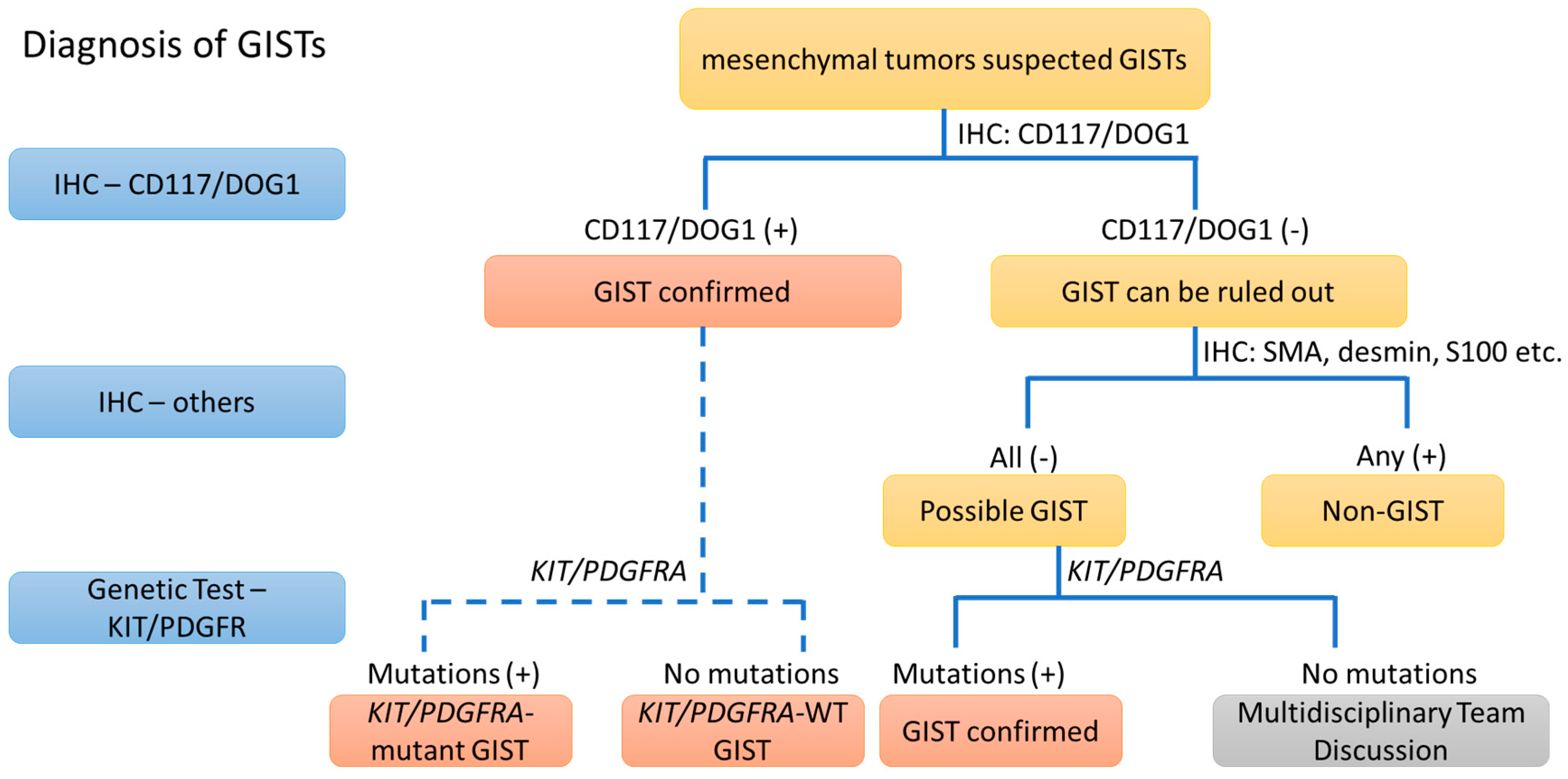
2. The Diagnosis of GIST from the IHC Point of View (>95%)
2.1. IHC of CD117
2.2. IHC of DOG1
2.3. IHC of PKCθ and Others
2.4. Conclusions of IHC
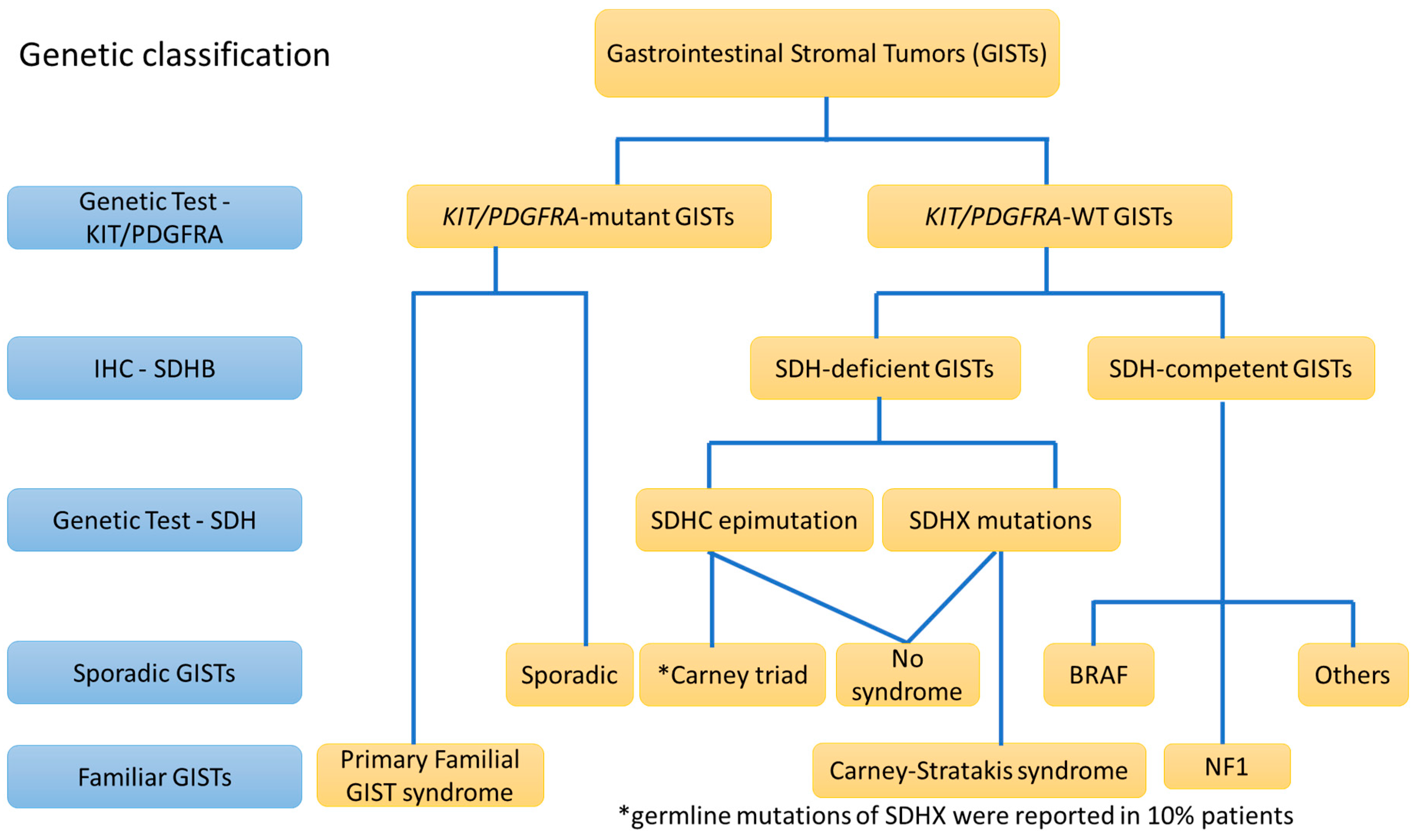
3. The Diagnostic View of Genetic Alterations in GISTs
3.1. KIT Mutations (80–85%)
3.2. PDGFRA Mutations (5–7%)
3.3. KIT/PDGFRA “Wild-Type (WT)” GISTs (5–10%)
4. CD117/DOG1-Positive GISTs with KIT/PDGFRA Mutations (80–85%)
5. CD117/DOG1-Negative GISTs with KIT/PDGFRA Mutations (<5%)
6. CD117/DOG1-Positive GISTs without KIT/PDGFRA Mutations (KIT/PDGFRA-WT) (5–10%)
6.1. CD117/DOG1-Positive and KIT/PDGFRA-WT GISTs: SDH-Deficient GISTs (>80% of KIT/PDGFRA-WT GISTs)
6.2. CD117/DOG1-Positive and KIT/PDGFRA-WT GISTs: SDH-Competent GISTs (<20% of KIT/PDGFRA-WT GISTs)
6.3. CD117/DOG1-Positive and KIT/PDGFRA-WT GISTs: GISTs in Neurofibromatosis Type I (NF1)
7. CD117/DOG1-Negative GISTs without KIT/PDGFRA Mutations (KIT/PDGFRA-WT) (<1%)
8. Germline Mutations vs. Sporadic Mutations
9. Conclusions
Funding
Conflicts of Interest
References
- Miettinen, M.; Sarlomo-Rikala, M.; Lasota, J. Gastrointestinal stromal tumors: Recent advances in understanding of their biology. Hum. Pathol. 1999, 30, 1213–1220. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Torihashi, S.; Ward, S.M.; Nishikawa, S.; Nishi, K.; Kobayashi, S.; Sanders, K.M. c-kit-dependent development of interstitial cells and electrical activity in the murine gastrointestinal tract. Cell Tissue Res. 1995, 280, 97–111. [Google Scholar] [PubMed]
- Torihashi, S.; Ward, S.M.; Sanders, K.M. Development of c-Kit-positive cells and the onset of electrical rhythmicity in murine small intestine. Gastroenterology 1997, 112, 144–155. [Google Scholar] [CrossRef]
- Ward, S.M.; Burns, A.J.; Torihashi, S.; Sanders, K.M. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J. Physiol. 1994, 480, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, J.D.; Thuneberg, L.; Kluppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/Kit Gene Required for Interstitial-Cells of Cajal and for Intestinal Pacemaker Activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef]
- Theou-Anton, N.; Tabone, S.; Brouty-Boye, D.; Saffroy, R.; Ronnstrand, L.; Lemoine, A.; Emile, J.F. Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/paracrine mechanism. Br. J. Cancer 2006, 94, 1180–1185. [Google Scholar] [CrossRef]
- Broudy, V.C. Stem cell factor and hematopoiesis. Blood 1997, 90, 1345–1364. [Google Scholar] [PubMed]
- Sarlomo-Rikala, M.; Kovatich, A.J.; Barusevicius, A.; Miettinen, M. CD117: A sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod. Pathol. 1998, 11, 728–734. [Google Scholar]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (GIPACT)-Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar]
- Sircar, K.; Hewlett, B.R.; Huizinga, J.D.; Chorneyko, K.; Berezin, I.; Riddell, R.H. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am. J. Surg. Pathol. 1999, 23, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Rankin, C.; Demetri, G.D.; Ryan, C.W.; von Mehren, M.; Benjamin, R.S.; Raymond, A.K.; Bramwell, V.H.C.; Baker, L.H.; Maki, R.G.; et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J. Clin. Oncol. 2008, 26, 626–632. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. New Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Yeh, C.N.; Cheng, C.T.; Wu, C.E.; Chiang, K.C.; Chen, T.W.; Wang, C.C.; Chen, J.S.; Yeh, T.S. Fractioned Dose Regimen of Sunitinib for Patients with Gastrointestinal Stromal Tumor: A Pharmacokinetic and Treatment Efficacy Study. Transl. Oncol. 2014, 7, 620–625. [Google Scholar] [CrossRef]
- Hsu, C.C.; Wu, C.E.; Chen, J.S.; Tseng, J.H.; Chiang, K.C.; Liu, Y.Y.; Tsai, C.Y.; Cheng, C.T.; Chen, T.W.; Jan, Y.Y.; et al. Imatinib Escalation or Sunitinib Treatment After First-line Imatinib in Metastatic Gastrointestinal Stromal Tumor Patients. Anticancer Res. 2014, 34, 5029–5036. [Google Scholar] [PubMed]
- Yeh, C.N.; Hwang, T.L.; Huang, C.S.; Lee, P.H.; Wu, C.W.; Chen-Guo, K.; Jan, Y.Y.; Chen, M.F.; Soc, T.S. Clinical practice guidelines for patients with gastrointestinal stromal tumor in Taiwan. World J. Surg. Oncol. 2012, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Yeh, C.N.; Cheng, C.T.; Chen, T.W.; Rau, K.M.; Jan, Y.Y.; Chen, M.F. Sunitinib for Taiwanese patients with gastrointestinal stromal tumor after imatinib treatment failure or intolerance. World J. Gastroentero. 2011, 17, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Sobin, L.H.; Sarlomo-Rikala, M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod. Pathol. 2000, 13, 1134–1142. [Google Scholar] [CrossRef]
- Miettinen, M.; Sarlomo-Rikala, M.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors and leiomyosarcomas in the colon: A clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases. Am. J. Surg. Pathol. 2000, 24, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Moskaluk, C.A.; Tian, Q.; Marshall, C.R.; Rumpel, C.A.; Franquemont, D.W.; Frierson, H.F. Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18, 1897–1902. [Google Scholar] [CrossRef]
- Taniguchi, M.; Nishida, T.; Hirota, S.; Isozaki, K.; Ito, T.; Nomura, T.; Matsuda, H.; Kitamura, Y. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res. 1999, 59, 4297–4300. [Google Scholar] [PubMed]
- Miettinen, M.; Furlong, M.; Sarlomo-Rikala, M.; Burke, A.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the rectum and anus-A clinicopathologic, immunohistochemical, and molecular genetic study of 144 cases. Am. J. Surg. Pathol. 2001, 25, 1121–1133. [Google Scholar] [CrossRef]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A.; et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar]
- Miettinen, M.; Wang, Z.F.; Lasota, J. DOG1 Antibody in the Differential Diagnosis of Gastrointestinal Stromal Tumors A Study of 1840 Cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef]
- Edris, B.; Willingham, S.B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Muhlenberg, T.; Montgomery, K.D.; Contreras-Trujillo, H.; Czechowicz, A.; Fletcher, J.A.; et al. Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 3501–3506. [Google Scholar] [CrossRef]
- London, C.A.; Gardner, H.L.; Rippy, S.; Post, G.; La Perle, K.; Crew, L.; Lopresti-Morrow, L.; Garton, A.J.; McMahon, G.; LaVallee, T.M.; et al. KTN0158, a Humanized Anti-KIT Monoclonal Antibody, Demonstrates Biologic Activity against both Normal and Malignant Canine Mast Cells. Clin. Cancer Res. 2017, 23, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- West, R.B.; Corless, C.L.; Chen, X.; Rubin, B.P.; Subramanian, S.; Montgomery, K.; Zhu, S.; Ball, C.A.; Nielsen, T.O.; Patel, R.; et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am. J. Pathol. 2004, 165, 107–113. [Google Scholar] [CrossRef]
- Qu, Z.Q.; Yao, W.C.; Yao, R.Y.; Liu, X.P.; Yu, K.; Hartzell, C. The Ca2+-activated Cl− channel, ANO1 (TMEM16A), is a double-edged sword in cell proliferation and tumorigenesis. Cancer Med. 2014, 3, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Stanich, J.E.; Gibbons, S.J.; Eisenman, S.T.; Bardsley, M.R.; Rock, J.R.; Harfe, B.D.; Ordog, T.; Farrugia, G. Ano1 as a regulator of proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1044–G1051. [Google Scholar] [CrossRef]
- Simon, S.; Grabellus, F.; Ferrera, L.; Galietta, L.; Schwindenhammer, B.; Muhlenberg, T.; Taeger, G.; Eilers, G.; Treckmann, J.; Breitenbuecher, F.; et al. DOG1 regulates growth and IGFBP5 in gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Akcakaya, P.; Berglund, D.; Karlsson, F.; Vukojevic, V.; Lee, L.; Bogdanovic, D.; Lui, W.O.; Larsson, C.; Zedenius, J.; et al. Functional role of the Ca(2)(+)-activated Cl(-) channel DOG1/TMEM16A in gastrointestinal stromal tumor cells. Exp. Cell Res. 2014, 326, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.F.; West, R.B.; Bacchi, L.M.; van de Rijn, M.; Bacchi, C.E. DOG1 for the Diagnosis of Gastrointestinal Stromal Tumor (GIST): Comparison Between 2 Different Antibodies. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 333–337. [Google Scholar] [CrossRef]
- Poole, D.P.; Van Nguyen, T.; Kawai, M.; Furness, J.B. Protein kinases expressed by interstitial cells of Cajal. Histochem. Cell Biol. 2004, 121, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.P.; Hunne, B.; Robbins, H.L.; Furness, J.B. Protein kinase C isoforms in the enteric nervous system. Histochem. Cell Biol. 2003, 120, 51–61. [Google Scholar] [CrossRef]
- Allander, S.V.; Nupponen, N.N.; Ringner, M.; Hostetter, G.; Maher, G.W.; Goldberger, N.; Chen, Y.D.; Carpten, J.; Elkahloun, A.G.; Meltzer, P.S. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res. 2001, 61, 8624–8628. [Google Scholar]
- Nielsen, T.O.; West, R.B.; Linn, S.C.; Alter, O.; Knowling, M.A.; O’Connell, J.X.; Zhu, S.; Fero, M.; Sherlock, G.; Pollack, J.R.; et al. Molecular characterisation of soft tissue tumours: A gene expression study. Lancet 2002, 359, 1301–1307. [Google Scholar] [CrossRef]
- Motegi, A.; Sakurai, S.; Nakayama, H.; Sano, T.; Oyama, T.; Nakajima, T. PKC theta, a novel immunohistochemical marker for gastrointestinal stromal tumors (GIST), especially useful for identifying KIT-negative tumors. Pathol. Int. 2005, 55, 106–112. [Google Scholar] [CrossRef]
- Blay, P.; Astudillo, A.; Buesa, J.M.; Campo, E.; Abad, M.; Garcia-Garcia, J.; Miquel, R.; Marco, V.; Sierra, M.; Losa, R.; et al. Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin. Cancer Res. 2004, 10, 4089–4095. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Srivastava, A.; Kim, Y.E.; Park, H.J.; Park, C.K.; Sohn, T.S.; Kim, S.; Kang, D.Y.; Kim, K.M. DOG1 and PKC-theta are useful in the diagnosis of KIT-negative gastrointestinal stromal tumors. Mod. Pathol. 2011, 24, 866–875. [Google Scholar] [CrossRef]
- Lee, H.E.; Kim, M.A.; Lee, H.S.; Lee, B.L.; Kim, W.H. Characteristics of KIT-negative gastrointestinal stromal tumours and diagnostic utility of protein kinase C theta immunostaining. J. Clin. Pathol. 2008, 61, 722–729. [Google Scholar] [CrossRef]
- Tsujimura, T.; Makiishi-Shimobayashi, C.; Lundkvist, J.; Lendahl, U.; Nakasho, K.; Sugihara, A.; Iwasaki, T.; Mano, M.; Yamada, N.; Yamashita, K.; et al. Expression of the intermediate filament nestin in gastrointestinal stromal tumors and interstitial cells of Cajal. Am. J. Pathol. 2001, 158, 817–823. [Google Scholar] [CrossRef]
- Wang, J.; Cai, J.; Huang, Y.; Ke, Q.; Wu, B.; Wang, S.; Han, X.; Wang, T.; Wang, Y.; Li, W.; et al. Nestin regulates proliferation and invasion of gastrointestinal stromal tumor cells by altering mitochondrial dynamics. Oncogene 2016, 35, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Novelli, M.; Rossi, S.; Rodriguez-Justo, M.; Taniere, P.; Seddon, B.; Toffolatti, L.; Sartor, C.; Hogendoorn, P.C.W.; Sciot, R.; Van Glabbeke, M.; et al. DOG1 and CD117 are the antibodies of choice in the diagnosis of gastrointestinal stromal tumours. Histopathology 2010, 57, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Jasinski, M.; Sarlomo-Rikala, M.; Miettinen, M. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am. J. Pathol. 1999, 154, 53–60. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Lux, M.L.; Rubin, B.P.; Biase, T.L.; Chen, C.J.; Maclure, T.; Demetri, G.; Xiao, S.; Singer, S.; Fletcher, C.D.; Fletcher, J.A. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am. J. Pathol. 2000, 156, 791–795. [Google Scholar] [CrossRef]
- Yeh, C.N.; Chen, M.H.; Chen, Y.Y.; Yang, C.Y.; Yen, C.C.; Tzen, C.Y.; Chen, L.T.; Chen, J.S. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget 2017, 8, 44121–44130. [Google Scholar] [CrossRef]
- Yeh, C.N.; Chen, Y.Y.; Tseng, J.H.; Chen, J.S.; Chen, T.W.; Tsai, C.Y.; Cheng, C.T.; Jan, Y.Y.; Chen, M.F. Imatinib Mesylate for Patients with Recurrent or Metastatic Gastrointestinal Stromal Tumors Expressing KIT: A Decade Experience from Taiwan. Transl. Oncol. 2011, 4, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.N.; Chen, T.W.; Tseng, J.H.; Liu, Y.Y.; Wang, S.Y.; Tsai, C.Y.; Chiang, K.C.; Hwang, T.L.; Jan, Y.Y.; Chen, M.F. Surgical management in metastatic gastrointestinal stromal tumor (GIST) patients after imatinib mesylate treatment. J. Surg. Oncol. 2010, 102, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Y.; Li, J.; Sun, N.; Yuan, J.; Shen, L. Secondary mutations of c-KIT contribute to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med. Oncol. 2013, 30, 522. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar] [CrossRef]
- Medeiros, F.; Corless, C.L.; Duensing, A.; Hornick, J.L.; Oliveira, A.M.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D. KIT-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. Am. J. Surg. Pathol. 2004, 28, 889–894. [Google Scholar] [CrossRef]
- Nishida, T.; Shirao, K.; Sawaki, A.; Koseki, M.; Okamura, T.; Ohtsu, A.; Sugiyama, T.; Miyakawa, K.; Hirota, S. Efficacy and safety profile of imatinib mesylate (ST1571) in Japanese patients with advanced gastrointestinal stromal tumors: A phase II study (STI571B1202). Int. J. Clin. Oncol. 2008, 13, 244–251. [Google Scholar] [CrossRef]
- Ryu, M.H.; Kang, W.K.; Bang, Y.J.; Lee, K.H.; Shin, D.B.; Ryoo, B.Y.; Roh, J.K.; Kang, J.H.; Lee, H.; Kim, T.W.; et al. A prospective, multicenter, phase 2 study of imatinib mesylate in korean patients with metastatic or unresectable gastrointestinal stromal tumor. Oncology 2009, 76, 326–332. [Google Scholar] [CrossRef]
- Wagner, A.J.; Kindler, H.; Gelderblom, H.; Schoffski, P.; Bauer, S.; Hohenberger, P.; Kopp, H.G.; Lopez-Martin, J.A.; Peeters, M.; Reichardt, P.; et al. A phase II study of a human anti-PDGFRalpha monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann. Oncol. 2017, 28, 541–546. [Google Scholar] [CrossRef]
- Martin, J.; Poveda, A.; Llombart-Bosch, A.; Ramos, R.; Lopez-Guerrero, J.A.; Garcia del Muro, J.; Maurel, J.; Calabuig, S.; Gutierrez, A.; Gonzalez de Sande, J.L.; et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: A study by the Spanish Group for Sarcoma Research (GEIS). J. Clin. Oncol. 2005, 23, 6190–6198. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Rios-Moreno, M.J.; Jaramillo, S.; Gallardo, S.P.; Vallejo, A.; Mora, M.; Garcia-Escudero, A.; Amerigo, J.; Gonzalez-Campora, R. Gastrointestinal stromal tumors (GISTs): CD117, DOG-1 and PKC theta expression. Is there any advantage in using several markers? Pathol. Res. Pract. 2012, 208, 74–81. [Google Scholar] [CrossRef]
- Liegl, B.; Hornick, J.L.; Corless, C.L.; Fletcher, C.D.M. Monoclonal Antibody DOG1.1 Shows Higher Sensitivity Than KIT in the Diagnosis of Gastrointestinal Stromal Tumors, Including Unusual Subtypes. Am. J. Surg. Pathol. 2009, 33, 437–446. [Google Scholar] [CrossRef]
- Duensing, A.; Joseph, N.E.; Medeiros, F.; Smith, F.; Hornick, J.L.; Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Fletcher, C.D.; Fletcher, J.A. Protein Kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res. 2004, 64, 5127–5131. [Google Scholar] [CrossRef]
- Emile, J.F.; Theou, N.; Tabone, S.; Cortez, A.; Terrier, P.; Chaumette, M.T.; Julie, C.; Bertheau, P.; Lavergne-Slove, A.; Donadieu, J.; et al. Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin. Gastroenterol. Hepatol. 2004, 2, 597–605. [Google Scholar] [CrossRef]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef]
- Mussi, C.; Schildhaus, H.U.; Gronchi, A.; Wardelmann, E.; Hohenberger, P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin. Cancer Res. 2008, 14, 4550–4555. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tobo, T.; Nakamori, M.; Imamura, M.; Kojima, A.; Oda, Y.; Nakamura, N.; Takahira, T.; Yao, T.; Tsuneyoshi, M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J. Cancer Res. Clin. 2009, 135, 791–798. [Google Scholar] [CrossRef]
- Scarpa, M.; Bertin, M.; Ruffolo, C.; Polese, L.; D’Amico, D.F.; Angriman, I. A systematic review on the clinical diagnosis of gastrointestinal stromal tumors. J. Surg. Oncol. 2008, 98, 384–392. [Google Scholar] [CrossRef]
- Janeway, K.A.; Pappo, A. Treatment Guidelines for Gastrointestinal Stromal Tumors in Children and Young Adults. J. Pediatr. Hematol./Oncol. 2012, 34, S69–S72. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J.; Sobin, L.H. Gastrointestinal stromal tumors of the stomach in children and young adults-A clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am. J. Surg. Pathol. 2005, 29, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Laquaglia, M.P.; Ustun, B.; Guo, T.; Wong, G.C.; Socci, N.D.; Maki, R.G.; DeMatteo, R.P.; Besmer, P.; Antonescu, C.R. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin. Cancer Res. 2008, 14, 3204–3215. [Google Scholar] [CrossRef]
- Call, J.; Walentas, C.D.; Eickhoff, J.C.; Scherzer, N. Survival of gastrointestinal stromal tumor patients in the imatinib era: Life raft group observational registry. BMC Cancer 2012, 12, 90. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nose, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Pappo, A.S.; Janeway, K.A. Pediatric gastrointestinal stromal tumors. Hematol. Oncol. Clin. N. Am. 2009, 23, 15–34. [Google Scholar] [CrossRef]
- Gill, A.J.; Chou, A.; Vilain, R.; Clarkson, A.; Lui, M.; Jin, R.; Tobias, V.; Samra, J.; Goldstein, D.; Smith, C.; et al. Immunohistochemistry for SDHB Divides Gastrointestinal Stromal Tumors (GISTs) into 2 Distinct Types. Am. J. Surg. Pathol. 2010, 34, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Wang, Z.F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate Dehydrogenase-Deficient GISTs: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 66 Gastric GISTs With Predilection to Young Age. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, A.S.; Bayley, J.P. The role of complex II in disease. Biochim. Biophys. Acta 2013, 1827, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmess, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Urbini, M.; Astolfi, A.; Indio, V.; Heinrich, M.C.; Corless, C.L.; Nannini, M.; Ravegnini, G.; Biasco, G.; Pantaleo, M.A. SDHC methylation in gastrointestinal stromal tumors (GIST): A case report. BMC Med. Genet. 2015, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A.; Sheps, S.G.; Go, V.L.; Gordon, H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N. Engl. J. Med. 1977, 296, 1517–1518. [Google Scholar] [CrossRef]
- Carney, J.A.; Stratakis, C.A. Familial paraganglioma and gastric stromal sarcoma: A new syndrome distinct from the Carney triad. Am. J. Med. Genet. 2002, 108, 132–139. [Google Scholar] [CrossRef]
- Gaal, J.; Stratakis, C.A.; Carney, J.A.; Ball, E.R.; Korpershoek, E.; Lodish, M.B.; Levy, I.; Xekouki, P.; van Nederveen, F.H.; den Bakker, M.A.; et al. SDHB immunohistochemistry: A useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod. Pathol. 2011, 24, 147–151. [Google Scholar] [CrossRef]
- Boikos, S.A.; Xekouki, P.; Fumagalli, E.; Faucz, F.R.; Raygada, M.; Szarek, E.; Ball, E.; Kim, S.Y.; Miettinen, M.; Helman, L.J.; et al. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur. J. Hum. Genet. 2016, 24, 569–573. [Google Scholar] [CrossRef]
- Killian, J.K.; Miettinen, M.; Walker, R.L.; Wang, Y.H.; Zhu, Y.J.; Waterfall, J.J.; Noyes, N.; Retnakumar, P.; Yang, Z.M.; Smith, W.I.; et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Lasota, J.; Wang, Z.; Kim, S.Y.; Helman, L.; Miettinen, M. Expression of the receptor for type i insulin-like growth factor (IGF1R) in gastrointestinal stromal tumors: An immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am. J. Surg. Pathol. 2013, 37, 114–119. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)-a review. Int. J. Biochem. Cell Biol. 2014, 53, 514–519. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.F.; Bringuier, P.P.; Scoazec, J.Y.; Coindre, J.M. BRAF mutation status in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef]
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF mutant gastrointestinal stromal tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef]
- Corless, C.L. Gastrointestinal stromal tumors: What do we know now? Mod. Pathol. 2014, 27, S1–S16. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, H.; Hidaka, E.; Ota, H.; Minami, S.; Kajiyama, M.; Kuraishi, A.; Mori, H.; Matsuda, Y.; Wada, S.; Sodeyama, H.; et al. Familial gastrointestinal stromal tumor with hyperpigmentation: Association with a germline mutation of the c-kit gene. Gastroenterology 2001, 120, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Hirota, S.; Okazaki, T.; Kitamura, Y.; O’Brien, P.; Kapusta, L.; Dardick, I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of cajal is germline mutation of the c-kit gene. Am. J. Surg. Pathol. 2000, 24, 326–327. [Google Scholar] [CrossRef] [PubMed]
- Beghini, A.; Tibiletti, M.; Roversi, G.; Chiaravalli, A.; Serio, G.; Capella, C.; Larizza, L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001, 92, 657–662. [Google Scholar] [CrossRef]
- Chompret, A.; Kannengiesser, C.; Barrois, M.; Terrier, P.; Dahan, P.; Tursz, T.; Lenoir, G.M.; Bressac-de Paillerets, B. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004, 126, 318–321. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Cools, J.; Debiec-Rychter, M.; Brems, H.; Mentens, N.; Sciot, R.; Himpens, J.; De Wever, I.; Schoffski, P.; Marynen, P.; et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology 2006, 131, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Pasini, B.; Matyakhina, L.; Bei, T.; Muchow, M.; Boikos, S.; Ferrando, B.; Carney, J.A.; Stratakis, C.A. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: A case associated with a germline V561D defect. J. Clin. Endocr. Metab. 2007, 92, 3728–3732. [Google Scholar] [CrossRef] [PubMed]
- Ricci, R.; Martini, M.; Cenci, T.; Carbone, A.; Lanza, P.; Biondi, A.; Rindi, G.; Cassano, A.; Larghi, A.; Persiani, R.; et al. PDGFRA-mutant syndrome. Mod. Pathol. 2015, 28, 954–964. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-E.; Tzen, C.-Y.; Wang, S.-Y.; Yeh, C.-N. Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View. Cancers 2019, 11, 679. https://doi.org/10.3390/cancers11050679
Wu C-E, Tzen C-Y, Wang S-Y, Yeh C-N. Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View. Cancers. 2019; 11(5):679. https://doi.org/10.3390/cancers11050679
Chicago/Turabian StyleWu, Chiao-En, Chin-Yuan Tzen, Shang-Yu Wang, and Chun-Nan Yeh. 2019. "Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View" Cancers 11, no. 5: 679. https://doi.org/10.3390/cancers11050679
APA StyleWu, C.-E., Tzen, C.-Y., Wang, S.-Y., & Yeh, C.-N. (2019). Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View. Cancers, 11(5), 679. https://doi.org/10.3390/cancers11050679