The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Use of Cysteine
3. Regulation of Cysteine Metabolism
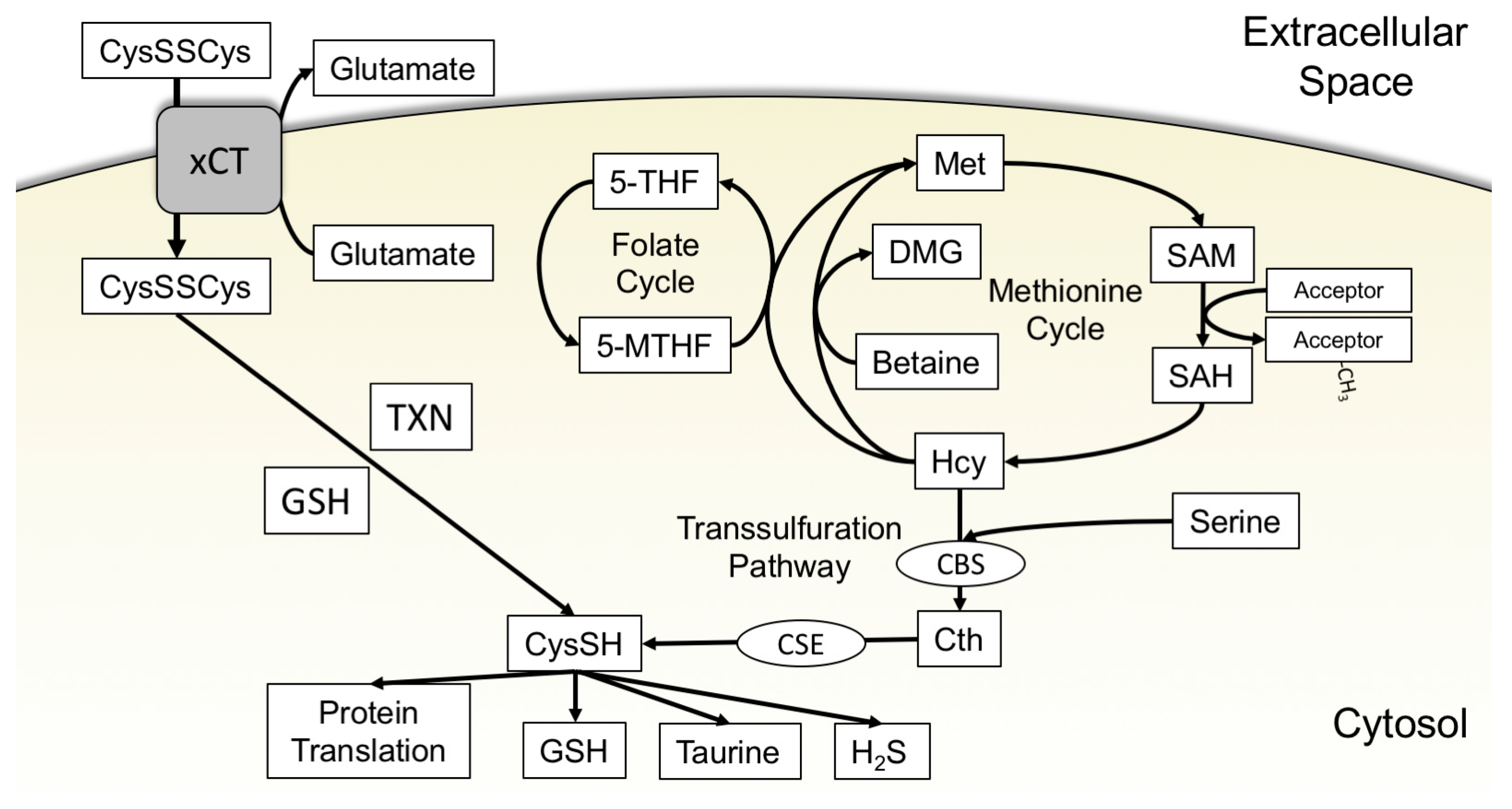
3.1. xCT Uptake of Cystine
Other Cystine Transporters
3.2. The Transsulfuration Pathway and de Novo Cysteine Synthesis
3.3. Cysteine-Specific Transporters
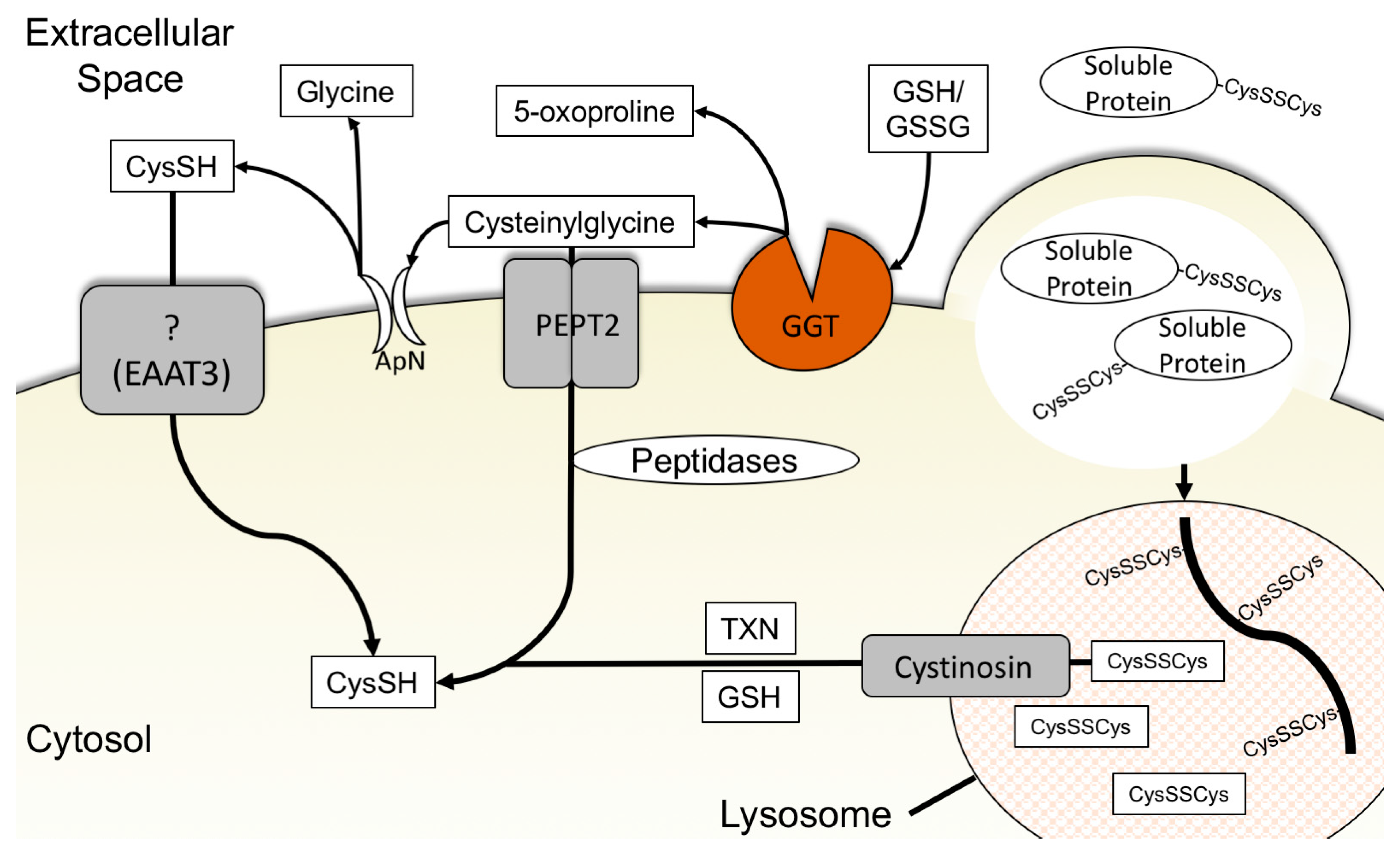
3.4. Glutathione Degradation and Cysteine Salvage
3.5. Macropinocytosis, Protein Scavenging, and Lysosomal Cyst(e)ine Transport
4. Conclusions and Remaining Questions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef]
- Narta, U.K.; Kanwar, S.S.; Azmi, W. Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. Hematol. 2007, 61, 208–221. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Ghisolfi, L.; Geck, R.C.; Asara, J.M.; Toker, A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal. 2017, 10, eaao6604. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 2004, 24, 539–577. [Google Scholar] [CrossRef]
- Bak, D.W.; Bechtel, T.J.; Falco, J.A.; Weerapana, E. Cysteine reactivity across the subcellular universe. Curr. Opin. Chem. Biol. 2019, 48, 96–105. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The Quantitatively Important Relationship between Homocysteine Metabolism and Glutathione Synthesis by the Transsulfuration Pathway and Its Regulation by Redox Changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276. [Google Scholar] [CrossRef]
- Hanigan, M.H.; Ricketts, W.A. Extracellular glutathione is a source of cysteine for cells that express gamma.-glutamyl transpeptidase. Biochemistry 1993, 32, 6302–6306. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2016, 23, 235. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2007, 27, 1618. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cysteine is not a substrate but a specific modulator of human ASCT2 (SLC1A5) transporter. FEBS Lett. 2015, 589, 3617–3623. [Google Scholar] [CrossRef]
- Kang, Y.P.; Torrente, L.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. bioRxiv 2018, 459602. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef] [PubMed]
- Poursaitidis, I.; Wang, X.; Crighton, T.; Labuschagne, C.; Mason, D.; Cramer, S.L.; Triplett, K.; Roy, R.; Pardo, O.E.; Seckl, M.J.; et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 2017, 18, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e127. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Dayal, S.; Stabler, S.; Zhou, Y.; Wang, H.; Lentz, S.R.; Banerjee, R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R39–R46. [Google Scholar] [CrossRef]
- Brigham, M.P.; Stein, W.H.; Moore, S. THE concentrations of cysteine and cystine in human blood plasma. J. Clin. Investig. 1960, 39, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 2017, 6, e27713. [Google Scholar] [CrossRef]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6, e28083. [Google Scholar] [CrossRef]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Forde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef] [PubMed]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016, 76, 62. [Google Scholar] [CrossRef] [PubMed]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased Expression of System Xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol. Cancer Res. 2016, 14, 1229. [Google Scholar] [CrossRef] [PubMed]
- De la Ballina, L.R.; Cano-Crespo, S.; Gonzalez-Munoz, E.; Bial, S.; Estrach, S.; Cailleteau, L.; Tissot, F.; Daniel, H.; Zorzano, A.; Ginsberg, M.H.; et al. Amino Acid Transport Associated to Cluster of Differentiation 98 Heavy Chain (CD98hc) Is at the Cross-road of Oxidative Stress and Amino Acid Availability. J. Biol. Chem. 2016, 291, 9700–9711. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.-I.; Moriguchi, T.; et al. Redox Imbalance in Cystine/Glutamate Transporter-deficient Mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the Cystine/Glutamate Antiporter System x−c in the Brain, Kidney, and Duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef]
- Siska, P.J.; Kim, B.; Ji, X.; Hoeksema, M.D.; Massion, P.P.; Beckermann, K.E.; Wu, J.; Chi, J.-T.; Hong, J.; Rathmell, J.C. Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes. J. Immunol. Methods 2016, 438, 51–58. [Google Scholar] [CrossRef]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef]
- Martin, L.; Gardner, L.B. Stress-induced inhibition of nonsense-mediated RNA decay regulates intracellular cystine transport and intracellular glutathione through regulation of the cystine/glutamate exchanger SLC7A11. Oncogene 2014, 34, 4211. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-Dependent Upregulation of xCT Modulates the Sensitivity of T24 Bladder Carcinoma Cells to Proteasome Inhibition. Mol. Cell. Biol. 2014, 34, 3421. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Somers, J.; Pöyry, T.; Willis, A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374. [Google Scholar] [CrossRef] [PubMed]
- Siu, F.; Bain, P.J.; LeBlanc-Chaffin, R.; Chen, H.; Kilberg, M.S. ATF4 Is a Mediator of the Nutrient-sensing Response Pathway That Activates the Human Asparagine Synthetase Gene. J. Biol. Chem. 2002, 277, 24120–24127. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.R.; Ong, S.-E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 2016, 5, e10575. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Basal Levels of eIF2α Phosphorylation Determine Cellular Antioxidant Status by Regulating ATF4 and xCT Expression. J. Biol. Chem. 2009, 284, 1106–1115. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 2017, 292, 19721–19732. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Sehm, T.; Fan, Z.; Ghoochani, A.; Rauh, M.; Engelhorn, T.; Minakaki, G.; Dorfler, A.; Klucken, J.; Buchfelder, M.; Eyupoglu, I.Y.; et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 2016, 7, 36021–36033. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e353. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of Sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Pras, E.; Raben, N.; Golomb, E.; Arber, N.; Aksentijevich, I.; Schapiro, J.M.; Harel, D.; Katz, G.; Liberman, U.; Pras, M.; et al. Mutations in the SLC3A1 transporter gene in cystinuria. Am. J. Hum. Genet. 1995, 56, 1297–1303. [Google Scholar]
- Jiang, Y.; Cao, Y.; Wang, Y.; Li, W.; Liu, X.; Lv, Y.; Li, X.; Mi, J. Cysteine transporter SLC3A1 promotes breast cancer tumorigenesis. Theranostics 2017, 7, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Inoue, H.; Tanaka, F.; Mimori, K.; Utsunomiya, T.; Sasaki, A.; Mori, M. Cancer stem cells in human gastrointestinal cancers. Hum. Cell 2006, 19, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 Mutations and Nrf2 Pathway Activation in Epithelial Ovarian Cancer. Cancer Res. 2011, 71, 5081. [Google Scholar] [CrossRef] [PubMed]
- Bartoccioni, P.; Rius, M.; Zorzano, A.; Palacin, M.; Chillaron, J. Distinct classes of trafficking rBAT mutants cause the type I cystinuria phenotype. Hum. Mol. Genet. 2008, 17, 1845–1854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pérez-Miguelsanz, J.; Vallecillo, N.; Garrido, F.; Reytor, E.; Pérez-Sala, D.; Pajares, M.A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1165–1182. [Google Scholar] [CrossRef]
- Rosado, J.O.; Salvador, M.; Bonatto, D. Importance of the trans-sulfuration pathway in cancer prevention and promotion. Mol. Cell. Biochem. 2007, 301, 1–12. [Google Scholar] [CrossRef]
- Korendyaseva, T.K.; Kuvatov, D.N.; Volkov, V.A.; Martinov, M.V.; Vitvitsky, V.M.; Banerjee, R.; Ataullakhanov, F.I. An Allosteric Mechanism for Switching between Parallel Tracks in Mammalian Sulfur Metabolism. PLoS Comput. Biol. 2008, 4, e1000076. [Google Scholar] [CrossRef]
- Tamanna, N.; Mayengbam, S.; House, J.D.; Treberg, J.R. Methionine restriction leads to hyperhomocysteinemia and alters hepatic H2S production capacity in Fischer-344 rats. Mech. Ageing Dev. 2018, 176, 9–18. [Google Scholar] [CrossRef]
- Prudova, A.; Bauman, Z.; Braun, A.; Vitvitsky, V.; Lu, S.C.; Banerjee, R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Oliveriusová, J.; Kery, V.r.; Maclean, K.N.; Kraus, J.P. Deletion Mutagenesis of Human Cystathionine β-Synthase: Impact on activity, oligomeric status, ands-adenosylmethionine regulation. J. Biol. Chem. 2002, 277, 48386–48394. [Google Scholar] [CrossRef]
- Zou, C.-G.; Banerjee, R. Tumor Necrosis Factor-α-induced Targeted Proteolysis of Cystathionine β-Synthase Modulates Redox Homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, J.G.; Carlisle, R.E.; Jerome, D.E.; Mohammed-Ali, Z.; Jiang, H.; Yang, G.; Mani, S.; Garg, S.K.; Banerjee, R.; Kaufman, R.J.; et al. Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 2012, 287, 7603–7614. [Google Scholar] [CrossRef]
- Maclean, K.N.; Greiner, L.S.; Evans, J.R.; Sood, S.K.; Lhotak, S.; Markham, N.E.; Stabler, S.P.; Allen, R.H.; Austin, R.C.; Balasubramaniam, V.; et al. Cystathionine protects against endoplasmic reticulum stress-induced lipid accumulation, tissue injury, and apoptotic cell death. J. Biol. Chem. 2012, 287, 31994–32005. [Google Scholar] [CrossRef]
- Maclean, K.N.; Sikora, J.; Kožich, V.; Jiang, H.; Greiner, L.S.; Kraus, E.; Krijt, J.; Overdier, K.H.; Collard, R.; Brodsky, G.L.; et al. A novel transgenic mouse model of CBS-deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol. Genet. Metab. 2010, 101, 153–162. [Google Scholar] [CrossRef]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Transsulfuration in mammals. Microassays and tissue distributions of three enzymes of the pathway. J. Biol. Chem. 1965, 240, 4382–4392. [Google Scholar] [PubMed]
- Zhao, H.; Li, Q.; Wang, J.; Su, X.; Ng, K.M.; Qiu, T.; Shan, L.; Ling, Y.; Wang, L.; Cai, J.; et al. Frequent Epigenetic Silencing of the Folate-Metabolising Gene Cystathionine-Beta-Synthase in Gastrointestinal Cancer. PLoS ONE 2012, 7, e49683. [Google Scholar] [CrossRef]
- Fukagawa, N.K.; Yu, Y.M.; Young, V.R. Methionine and cysteine kinetics at different intakes of methionine and cysteine in elderly men and women. Am. J. Clin. Nutr. 1998, 68, 380–388. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Cochran, F.B.; Sweetman, L.; Schmidt, K.; Barsh, G.; Kraus, J.; Packman, S. Pyridoxine-unresponsive homocystinuria with an unusual clinical course. Am. J. Med. Genet. 1990, 35, 519–522. [Google Scholar] [CrossRef]
- Watanabe, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huff, A.M.; Spence, J.D.; Hegele, R.A. Single nucleotide polymorphism in CTH associated with variation in plasma homocysteine concentration. Clin. Genet. 2004, 65, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, T.J.; Hornstra, N.; van Hoogstraten, B.; van der Meulen, J. Homocysteine, a risk factor for coronary artery disease or not? A meta-analysis. Am. J. Cardiol. 2000, 86, 1005–1009. [Google Scholar] [CrossRef]
- Li, J.-G.; Barrero, C.; Gupta, S.; Kruger, W.D.; Merali, S.; Praticò, D. Homocysteine modulates 5-lipoxygenase expression level via DNA methylation. Aging Cell 2017, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wen, X.; Wu, W.; Xu, E.; Zhang, Y.; Cui, W. Homocysteine-related hTERT DNA demethylation contributes to shortened leukocyte telomere length in atherosclerosis. Atherosclerosis 2013, 231, 173–179. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Diff. 2015, 23, 270. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hong, S.J.; Park, J.H.; Park, S.Y.; Kim, S.W.; Cho, E.Y.; Do, I.G.; Joh, J.W.; Kim, D.S. Expression of cystathionine beta-synthase is downregulated in hepatocellular carcinoma and associated with poor prognosis. Oncol. Rep. 2009, 21, 1449–1454. [Google Scholar] [CrossRef]
- McIntyre, T.; Curthoys, N.P. Renal catabolism of glutathione. Characterization of a particulate rat renal dipeptidase that catalyzes the hydrolysis of cysteinylglycine. J. Biol. Chem. 1982, 257, 11915–11921. [Google Scholar] [PubMed]
- Bjorn-Yoshimoto, W.E.; Underhill, S.M. The importance of the excitatory amino acid transporter 3 (EAAT3). Neurochem. Int. 2016, 98, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Fairman, W.A.; Wadiche, J.I.; Murdoch, G.H.; Kavanaugh, M.P.; Amara, S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 1994, 14, 5559. [Google Scholar] [CrossRef]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine Transport through Excitatory Amino Acid Transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Nkabyo, Y.S.; Go, Y.-M.; Ziegler, T.R.; Jones, D.P. Extracellular cysteine/cystine redox regulates the p44/p42 MAPK pathway by metalloproteinase-dependent epidermal growth factor receptor signaling. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, G70–G78. [Google Scholar] [CrossRef]
- Mannery, Y.O.; Ziegler, T.R.; Jones, D.P. A Chemically Defined Diet With Insufficient Sulfur Amino Acids Induces Oxidation of Plasma Cysteine/Cystine and Glutathione/Glutathione Disulfide Redox State in Humans. FASEB J. 2007, 21, A697. [Google Scholar] [CrossRef]
- Lieberman, M.W.; Wiseman, A.L.; Shi, Z.Z.; Carter, B.Z.; Barrios, R.; Ou, C.N.; Chévez-Barrios, P.; Wang, Y.; Habib, G.M.; Goodman, J.C.; et al. Growth retardation and cysteine deficiency in gamma-glutamyl transpeptidase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 7923. [Google Scholar] [CrossRef] [PubMed]
- Michelet, F.; Gueguen, R.; Leroy, P.; Wellman, M.; Nicolas, A.; Siest, G. Blood and plasma glutathione measured in healthy subjects by HPLC: relation to sex, aging, biological variables, and life habits. Clin. Chem. 1995, 41, 1509–1517. [Google Scholar]
- Cantin, A.M.; North, S.L.; Hubbard, R.C.; Crystal, R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1987, 63, 152–157. [Google Scholar] [CrossRef]
- Abbott, W.A.; Bridges, R.J.; Meister, A. Extracellular metabolism of glutathione accounts for its disappearance from the basolateral circulation of the kidney. J. Biol. Chem. 1984, 259, 15393–15400. [Google Scholar]
- Hanigan, M.H. Chapter Three - Gamma-Glutamyl Transpeptidase: Redox Regulation and Drug Resistance. In Advances in Cancer Research; Townsend, D.M., Tew, K.D., Eds.; Academic Press: Cambridge, MA USA, 2014; Volume 122, pp. 103–141. [Google Scholar]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef]
- Frey, I.M.; Rubio-Aliaga, I.; Siewert, A.; Sailer, D.; Drobyshev, A.; Beckers, J.; de Angelis, M.H.; Aubert, J.; Hen, A.B.; Fiehn, O.; et al. Profiling at mRNA, protein, and metabolite levels reveals alterations in renal amino acid handling and glutathione metabolism in kidney tissue of Pept2−/− mice. Physiol. Genom. 2007, 28, 301–310. [Google Scholar] [CrossRef][Green Version]
- Gerber, M.A.; Thung, S.N. Enzyme patterns in human hepatocellular carcinoma. Am. J. Pathol. 1980, 98, 395–400. [Google Scholar] [CrossRef]
- Luo, M.; Sun, W.; Wu, C.; Zhang, L.; Liu, D.; Li, W.; Mei, Q.; Hu, G. High pretreatment serum gamma-glutamyl transpeptidase predicts an inferior outcome in nasopharyngeal carcinoma. Oncotarget 2017, 8, 67651–67662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gallagher, B.C.; Townsend, D.M.; Hanigan, M.H.; Gabarra, V. γ-glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatin in vivo. Carcinogenesis 1999, 20, 553–559. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef]
- Zille, M.; Kumar, A.; Kundu, N.; Bourassa, M.W.; Wong, V.S.C.; Willis, D.; Karuppagounder, S.S.; Ratan, R.R. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Wang, C.-K.; Yang, S.-C.; Hsu, S.-C.; Chang, F.-P.; Lin, Y.-T.; Chen, S.-F.; Cheng, C.-L.; Hsiao, M.; Lu, F.L.; Lu, J. CHAC2 is essential for self-renewal and glutathione maintenance in human embryonic stem cells. Free Radic. Biol. Med. 2017, 113, 439–451. [Google Scholar] [CrossRef]
- Goebel, G.; Berger, R.; Strasak, A.M.; Egle, D.; Muller-Holzner, E.; Schmidt, S.; Rainer, J.; Presul, E.; Parson, W.; Lang, S.; et al. Elevated mRNA expression of CHAC1 splicing variants is associated with poor outcome for breast and ovarian cancer patients. Br. J. cancer 2012, 106, 189–198. [Google Scholar] [CrossRef]
- Stehle, G.; Sinn, H.; Wunder, A.; Schrenk, H.H.; Stewart, J.C.; Hartung, G.; Maier-Borst, W.; Heene, D.L. Plasma protein (albumin) catabolism by the tumor itself--implications for tumor metabolism and the genesis of cachexia. Crit. Rev. Oncol. Hematol. 1997, 26, 77–100. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544. [Google Scholar] [CrossRef]
- Bloomfield, G.; Kay, R.R. Uses and abuses of macropinocytosis. J. Cell Sci. 2016, 129, 2697. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Araki, J.; King, B.; DeMatteo, R.G.; Thompson, C.B. Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proc. Natl. Acad. Sci. USA 2017, 114, E8628. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361. [Google Scholar] [CrossRef] [PubMed]
- Nofal, M.; Zhang, K.; Han, S.; Rabinowitz, J.D. mTOR Inhibition Restores Amino Acid Balance in Cells Dependent on Catabolism of Extracellular Protein. Mol. Cell 2017, 67, 936–946.e935. [Google Scholar] [CrossRef]
- Pisoni, R.L.; Acker, T.L.; Lisowski, K.M.; Lemons, R.M.; Thoene, J.G. A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J. Cell Biol. 1990, 110, 327. [Google Scholar] [CrossRef]
- Kalatzis, V.; Cherqui, S.; Antignac, C.; Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H+-driven lysosomal cystine transporter. EMBO J. 2001, 20, 5940. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; van den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 47. [Google Scholar] [CrossRef]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678. [Google Scholar] [CrossRef]
- Zhang, W.; Braun, A.; Bauman, Z.; Olteanu, H.; Madzelan, P.; Banerjee, R. Expression Profiling of Homocysteine Junction Enzymes in the NCI60 Panel of Human Cancer Cell Lines. Cancer Res. 2005, 65, 1554. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancer models reveals determinants of tumor nutrient availability. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272.e217. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.H.; Meier, T.; Issels, R.D.; Brielmeier, M.; Scheffer, B.; Bornkamm, G.W. Apoptosis in Burkitt lymphoma cells is prevented by promotion of cysteine uptake. Int. J. Cancer 1998, 75, 620–625. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. https://doi.org/10.3390/cancers11050678
Combs JA, DeNicola GM. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers. 2019; 11(5):678. https://doi.org/10.3390/cancers11050678
Chicago/Turabian StyleCombs, Joseph A., and Gina M. DeNicola. 2019. "The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival" Cancers 11, no. 5: 678. https://doi.org/10.3390/cancers11050678
APA StyleCombs, J. A., & DeNicola, G. M. (2019). The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers, 11(5), 678. https://doi.org/10.3390/cancers11050678