Effects of Copper Chelation on BRAFV600E Positive Colon Carcinoma Cells

 , , and
, , and 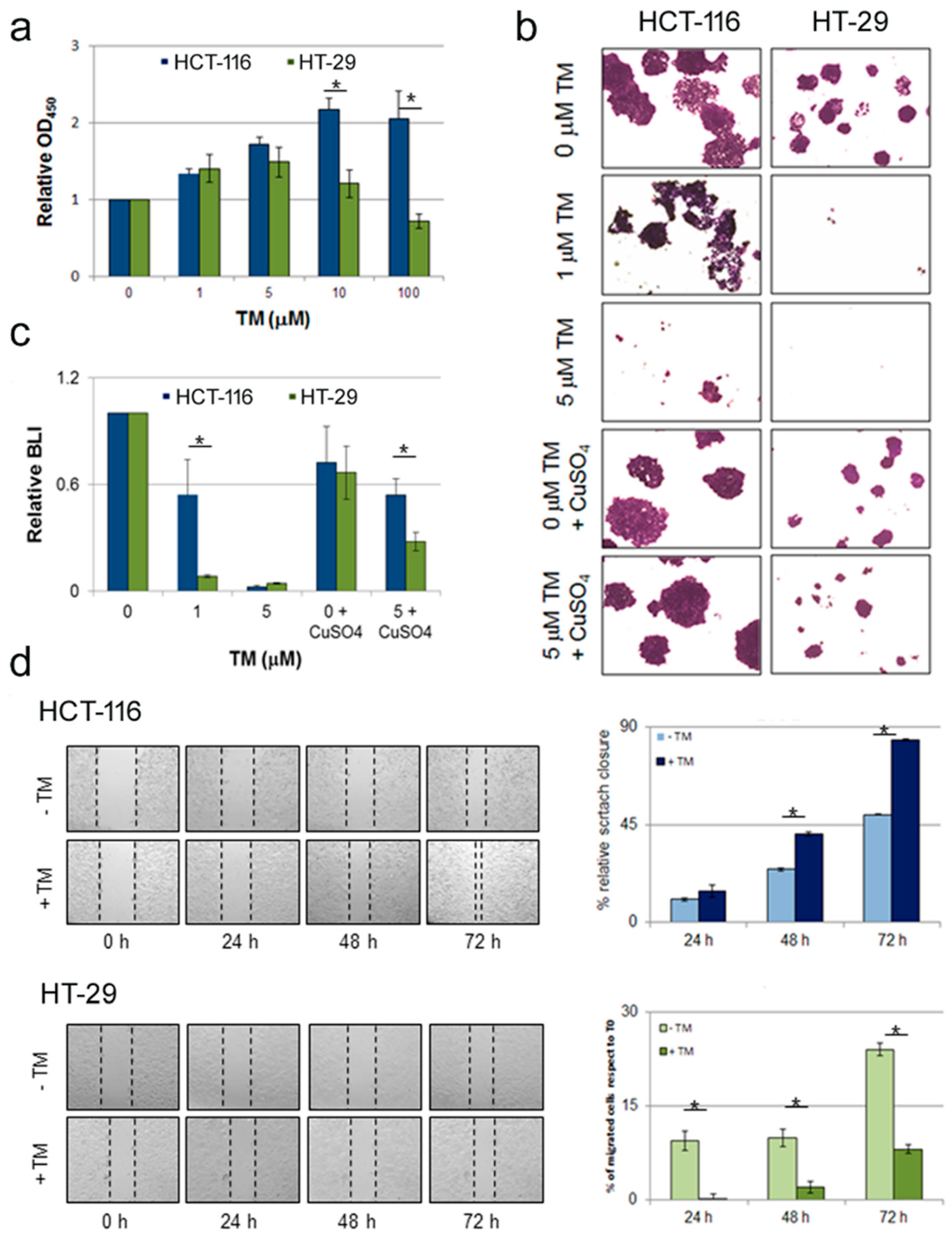{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Copper Modulation Differentially Affects Proliferation, Survival and Migration of Colon Cancer Cells Bearing BRAFV600E Mutation Compared to BRAFwt
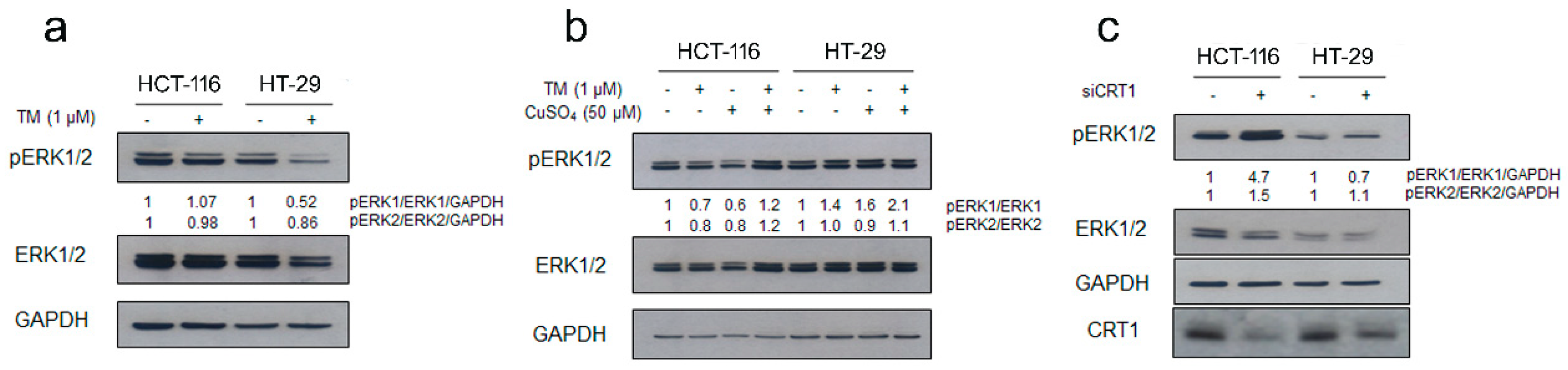
2.2. Copper Modulation Differentially Affects Phosphorylation of ERK1/2 in BRAFwt and BRAFV600E Colon Cancer Cells Lines
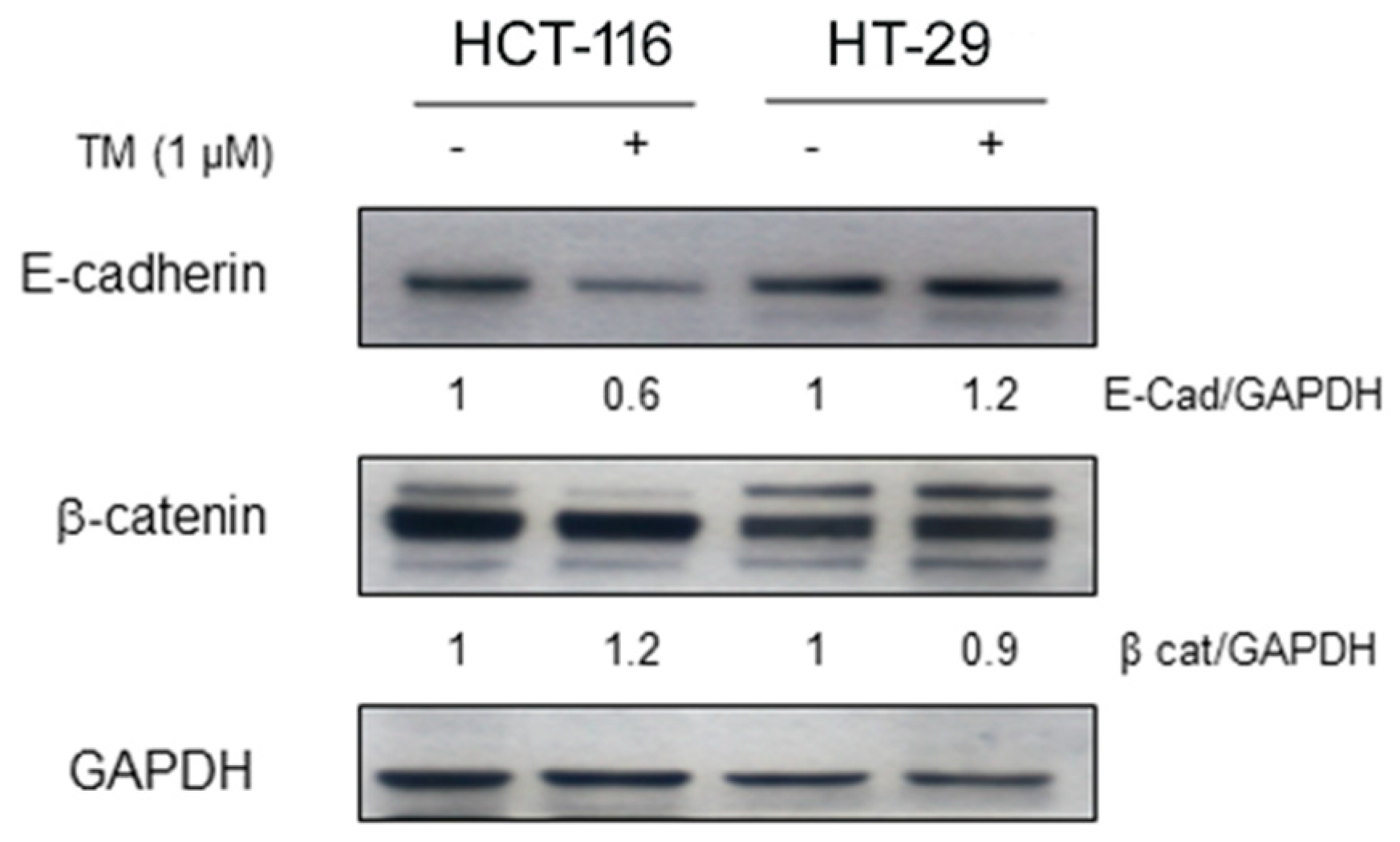
2.3. Copper Chelation Differently Affects E-Cadherin Expression in BRAFwt and BRAFV600E Colon Cancer Cell Lines
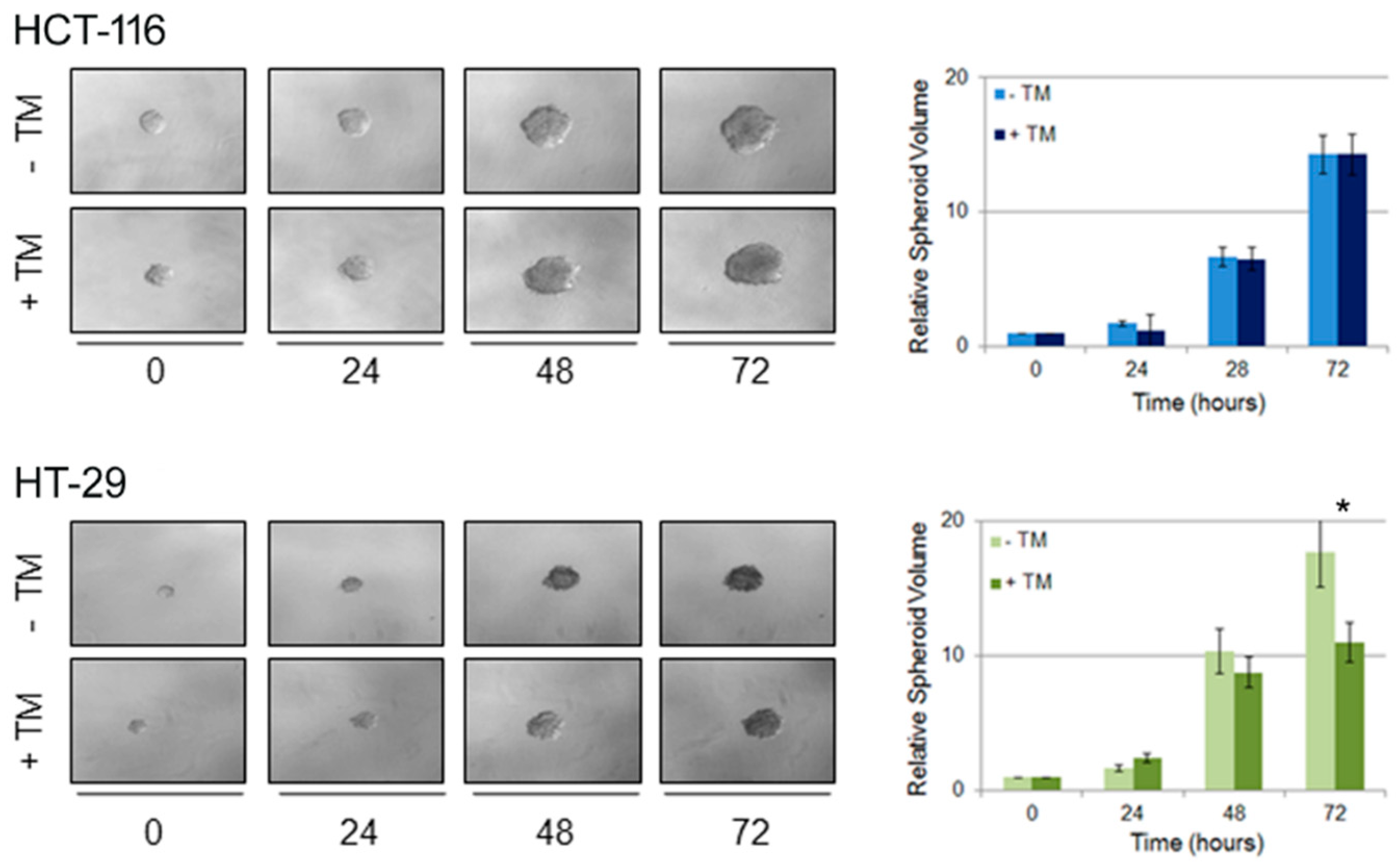
2.4. Copper Modulation Differentially Affects Proliferation of BRAFwt and BRAFV600E Colon Cancer Cells in a Three-Dimensional Tumor Spheroid Model
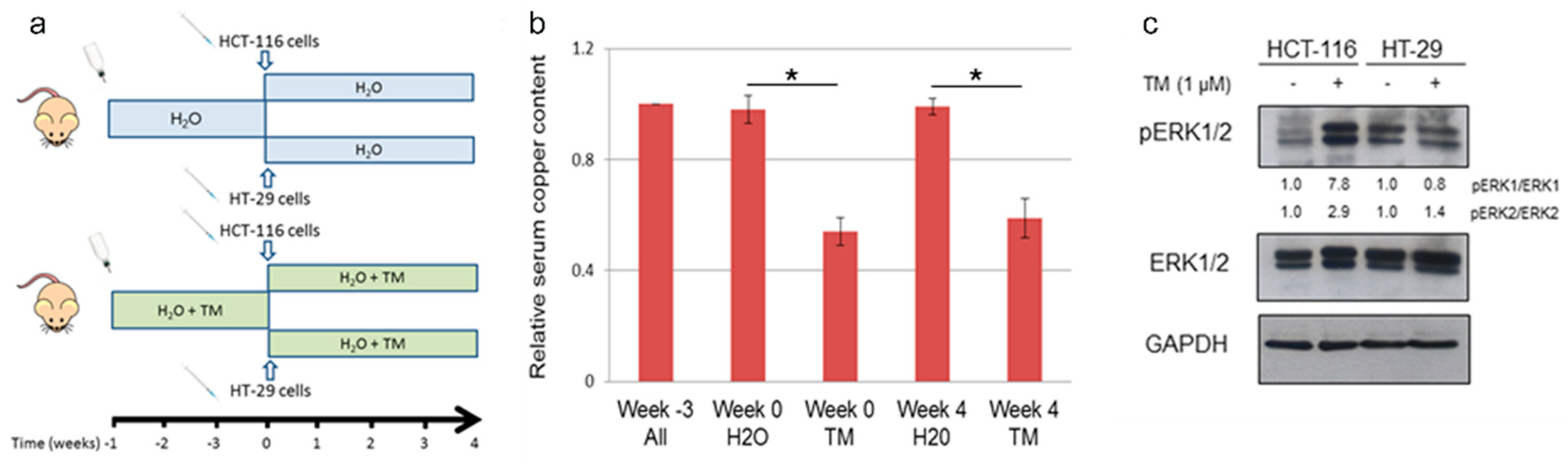
2.5. Copper Chelation Therapy Affects Human Tumor Growth in a Xenograft Model In Vivo
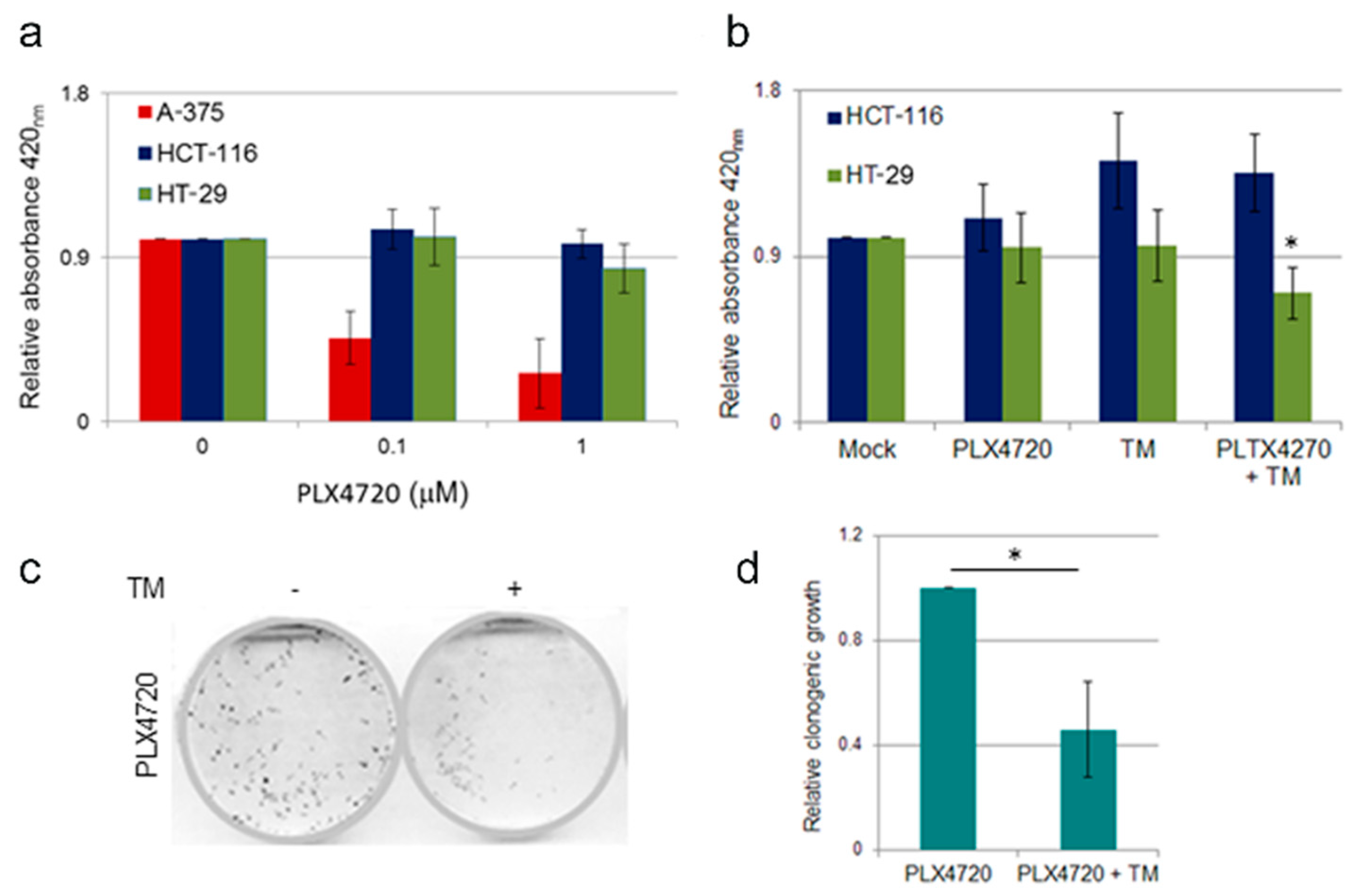
2.6. Effect of TM Treatment on BRAF Inhibitor Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Induction of BRAF Inhibition Resistance
4.3. Small Interfering RNA Transfection Procedure
4.4. In vitro Treatments and Cell Proliferation Assay
4.5. Colony Formation Assay
4.6. In vitro Scratch Assay
4.7. Immunoblotting
4.8. Experimental Animal Procedures
4.9. Optical Bioluminescence Imaging
4.10. 3D Tumor Spheroids Invasion Assay
4.11. Bioluminescence Analysis Using Copper Caged-Luciferin-1
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stintzing, S. Management of colorectal cancer. F1000Prime Rep. 2014, 6, 108. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Zhang, W. BRAF inhibitors: The current and the future. Curr. Opin. Pharmacol. 2015, 23, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B. New therapeutic strategies for BRAF mutant colorectal cancers. J. Gastrointest. Oncol. 2015, 6, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Ursem, C.; Atreya, C.E.; Van Loon, K. Emerging treatment options for BRAF-mutant colorectal cancer. Gastrointest Cancer 2018, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signaling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The role of copper in drug-resistant murine and human tumors. Biometals 2009, 22, 377–384. [Google Scholar] [CrossRef]
- Wu, T.; Sempos, C.T.; Freudenheim, J.L.; Muti, P.; Smit, E. Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults. Ann. Epidemiol. 2004, 14, 195–201. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ’Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Lowndes, S.A.; Harris, A.L. The role of copper in tumour angiogenesis. J. Mammary Gland Biol. Neoplasia 2005, 10, 299–310. [Google Scholar] [CrossRef]
- Medici, V.; Sturniolo, G.C. Tetrathiomolybdate, a copper chelator for the treatment of Wilson disease, pulmonary fibrosis and other indications. IDrugs 2008, 11, 592–606. [Google Scholar] [PubMed]
- Heffern, M.C.; Park, H.M.; Au-Yeung, H.Y.; Van de Bittner, G.C.; Ackerman, C.M.; Stahl, A.; Chang, C.J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2016, 113, 14219–14224. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frieboes, H.B.; Zheng, X.; Sun, C.H.; Tromberg, B.; Gatenby, R.; Cristini, V. An integrated computational/experimental model of tumor invasion. Cancer Res. 2006, 66, 1597–1604. [Google Scholar] [CrossRef]
- Zeng, C.; Hou, G.; Dick, R.; Brewer, G.J. Tetrathiomolybdate is partially protective against hyperglycemia in rodent models of diabetes. Exp. Biol. Med. (Maywood) 2008, 233, 1021–1025. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer. Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Clatworthy, S.A.S.; Cater, M.A. Copper complexes in cancer therapy. Met. Ions. Life Sci. 2018, 18. [Google Scholar] [CrossRef]
- Dou, Q.P. Repositioning the old, existing copper-binding drugs for cancer treatment. Clin. Exp. Pharmacol. 2012, 2, 1000e1102. [Google Scholar] [CrossRef]
- Wang, F.; Jiao, P.; Qi, M.; Frezza, M.; Dou, Q.P.; Yan, B. Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr. Med. Chem. 2010, 17, 2685–2698. [Google Scholar] [CrossRef]
- Goodman, V.L.; Brewer, G.J.; Merajver, S.D. Copper deficiency as an anti-cancer strategy. Endocr. Relat. Cancer 2004, 11, 255–263. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef]
- Daniel, K.G.; Chen, D.; Yan, B.; Dou, Q.P. Copper-binding compounds as proteasome inhibitors and apoptosis inducers in human cancer. Front. Biosci. 2007, 12, 135–144. [Google Scholar] [CrossRef]
- Barras, D.; Missiaglia, E.; Wirapati, P.; Sieber, O.M.; Jorissen, R.N.; Love, C.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E mutant colorectal cancer subtypes based on gene expression. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Fatfat, M.; Merhi, R.A.; Rahal, O.; Stoyanovsky, D.A.; Zaki, A.; Haidar, H.; Kagan, V.E.; Gali-Muhtasib, H.; Machaca, K. Copper chelation selectively kills colon cancer cells through redox cycling and generation of reactive oxygen species. BMC Cancer 2014, 14, 527. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Shukla, V.K.; Vaidya, M.P.; Roy, S.K.; Gupta, S. Serum and tissue trace elements in colorectal cancer. J. Surg. Oncol. 1993, 52, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.B.; Bhat, V.R.; Upadhyay, D.; Udupa, S.L. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J. Physiol. Pharmacol. 2003, 47, 108–110. [Google Scholar]
- Coates, R.J.; Weiss, N.S.; Daling, J.R.; Rettmer, R.L.; Warnick, G.R. Cancer risk in relation to serum copper levels. Cancer Res. 1989, 49, 4353–4356. [Google Scholar] [PubMed]
- Linder, M.C.; Moor, J.R.; Wright, K. Ceruloplasmin assays in diagnosis and treatment of human lung, breast, and gastrointestinal cancers. J. Natl. Cancer Inst. 1981, 67, 263–275. [Google Scholar] [CrossRef]
- Barresi, V.; Trovato-Salinaro, A.; Spampinato, G.; Musso, N.; Castorina, S.; Rizzarelli, E.; Condorelli, D.F. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1,SCO1, and COX11 in colorectal cancer. FEBS Open Bio. 2016, 6, 794–806. [Google Scholar] [CrossRef]
- Finney, L.; Vogt, S.; Fukai, T.; Glesne, D. Copper and angiogenesis: Unravelling a relationship key to cancer progression. Clin. Exp. Pharmacol. Physiol. 2009, 36, 88–94. [Google Scholar] [CrossRef]
- Antoniades, V.; Sioga, A.; Dietrich, E.M.; Meditskou, S.; Ekonomou, L.; Antoniades, K. Is copper chelation an effective anti-angiogenic strategy for cancer treatment? Med. Hypotheses 2013, 81, 1159–1163. [Google Scholar] [CrossRef]
- Goodman, V.L.; Brewer, G.J.; Merajver, S.D. Control of copper status for cancer therapy. Curr. Cancer Drug Targets 2005, 5, 543–549. [Google Scholar] [CrossRef]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the tumor microenvironment: A phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Jain, S.; Cohen, J.; Ward, M.M.; Kornhauser, N.; Chuang, E.; Cigler, T.; Moore, A.; Donovan, D.; Lam, C.; Cobham, M.V.; et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann. Oncol. 2013, 24, 1491–1498. [Google Scholar] [CrossRef]
- Gartner, E.M.; Griffith, K.A.; Pan, Q.; Brewer, G.J.; Henja, G.F.; Merajver, S.D.; Zalupski, M.M. A pilot trial of the anti-angiogenic copper lowering agent tetrathiomolybdate in combination with irinotecan, 5-flurouracil, and leucovorin for metastatic colorectal cancer. Invest. New Drugs 2009, 27, 159–165. [Google Scholar] [CrossRef]
- Redman, B.G.; Esper, P.; Pan, Q.; Dunn, R.L.; Hussain, H.K.; Chenevert, T.; Brewer, G.J.; Merajver, S.D. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. 2003, 9, 1666–1672. [Google Scholar]
- Henry, N.L.; Dunn, R.; Merjaver, S.; Pan, Q.; Pienta, K.J.; Brewer, G.; Smith, D.C. Phase II trial of copper depletion with tetrathiomolybdate as an antiangiogenesis strategy in patients with hormone-refractory prostate cancer. Oncology 2006, 71, 168–175. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin. Cancer Res. 2000, 6, 1–10. [Google Scholar]
- Schneider, B.J.; Lee, J.S.; Hayman, J.A.; Chang, A.C.; Orringer, M.B.; Pickens, A.; Pan, C.C.; Merajver, S.D.; Urba, S.G. Pre-operative chemoradiation followed by post-operative adjuvant therapy with tetrathiomolybdate, a novel copper chelator, for patients with resectable esophageal cancer. Invest. New Drugs 2013, 31, 435–442. [Google Scholar] [CrossRef]
- Brewer, G.J.; Merajver, S.D. Cancer therapy with tetrathiomolybdate: antiangiogenesis by lowering body copper—A review. Integr. Cancer Ther. 2002, 1, 327–337. [Google Scholar] [CrossRef]
- Khan, G.; Merajver, S. Copper chelation in cancer therapy using tetrathiomolybdate: an evolving paradigm. Expert Opin. Investig. Drugs 2009, 18, 541–548. [Google Scholar] [CrossRef]
- Sammons, S.; Brady, D.; Vahdat, L.; Salama, A.K. Copper suppression as cancer therapy: The rationale for copper chelating agents in BRAF V600 mutated melanoma. Melanoma Manag. 2016, 3, 207–216. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper chelation inhibits BRAF V600E-driven melanomagenesis and counters resistance to BRAF V600E and MEK1/2 inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Furukawa, T.; Komatsu, M.; Ikeda, R.; Tsujikawa, K.; Akiyama, S. Copper transport systems are involved in multidrug resistance and drug transport. Curr. Med. Chem. 2008, 15, 3268–3278. [Google Scholar] [CrossRef]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef]
- Faghfuri, E.; Nikfar, S.; Niaz, K.; Faramarzi, M.A.; Abdollahi, M. Mitogen-activated protein kinase (MEK) inhibitors to treat melanoma alone or in combination with other kinase inhibitors. Expert Opin. Drug Metab. Toxicol. 2018, 14, 317–330. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Sini, M.C.; Doneddu, V.; Paliogiannis, P.; Casula, M.; Colombino, M.; Manca, A.; Botti, G.; Ascierto, P.A.; Lissia, A.; Cossu, A.; et al. Genetic alterations in main candidate genes during melanoma progression. Oncotarget 2018, 9, 8531–8541. [Google Scholar] [CrossRef]
- Cosmic Cell Lines Project HCT-116 Mutations Cell Line Synopsis. Available online: http://www.webcitation.org/788mDTpZ8 (accessed on 05 May 2019).
- Cosmic Cell Lines Project HT-29 Mutations Cell Line Synopsis. Available online: http://www.webcitation.org/788l6jCw9 (accessed on 05 May 2019).
- Cosmic Cell Lines Project A-375 Mutations Cell Line Synopsis. Available online: http://www.webcitation.org/788on5GTQ (accessed on 05 May 2019).
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Panzini, G.; Lorenzini, R.N. Animal experimentation in Italy. Legislation and the authorization of research protocols. Ann Ist Super Sanita 2004, 40, 205–210. [Google Scholar]
- Cox, C.; Merajver, S.D.; Yoo, S.; Dick, R.D.; Brewer, G.J.; Lee, J.S.; Teknos, T.N. Inhibition of the growth of squamous cell carcinoma by tetrathiomolybdate-induced copper suppression in a murine model. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 781–785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, M.; Casio, M.; Range, D.E.; Sosa, J.A.; Counter, C.M. Copper chelation as targeted therapy in a mouse model of oncogenic BRAF-driven papillary thyroid cancer. Clin. Cancer Res. 2018, 24, 4271–4281. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, G.; Verdina, A.; Gatti, V.; Virdia, I.; Toietta, G.; Todaro, M.; Stassi, G.; Soddu, S. Apoptosis induced by a HIPK2 full-length-specific siRNA is due to off-target effects rather than prevalence of HIPK2-Delta e8 isoform. Oncotarget 2016, 7, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, G.; Gentile, A.; Antonini, A.; Truffa, S.; Piaggio, G.; Capogrossi, M.; Toietta, G. Analysis of biodistribution and engraftment into the liver of genetically modified mesenchymal stromal cells derived from adipose tissue. Cell Transplant. 2012, 21, 1997–2008. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldari, S.; Di Rocco, G.; Heffern, M.C.; Su, T.A.; Chang, C.J.; Toietta, G. Effects of Copper Chelation on BRAFV600E Positive Colon Carcinoma Cells. Cancers 2019, 11, 659. https://doi.org/10.3390/cancers11050659
Baldari S, Di Rocco G, Heffern MC, Su TA, Chang CJ, Toietta G. Effects of Copper Chelation on BRAFV600E Positive Colon Carcinoma Cells. Cancers. 2019; 11(5):659. https://doi.org/10.3390/cancers11050659
Chicago/Turabian StyleBaldari, Silvia, Giuliana Di Rocco, Marie C. Heffern, Timothy A. Su, Christopher J. Chang, and Gabriele Toietta. 2019. "Effects of Copper Chelation on BRAFV600E Positive Colon Carcinoma Cells" Cancers 11, no. 5: 659. https://doi.org/10.3390/cancers11050659
APA StyleBaldari, S., Di Rocco, G., Heffern, M. C., Su, T. A., Chang, C. J., & Toietta, G. (2019). Effects of Copper Chelation on BRAFV600E Positive Colon Carcinoma Cells. Cancers, 11(5), 659. https://doi.org/10.3390/cancers11050659