Targeting ATG4 in Cancer Therapy

 , ,
, , 
Abstract
1. Introduction
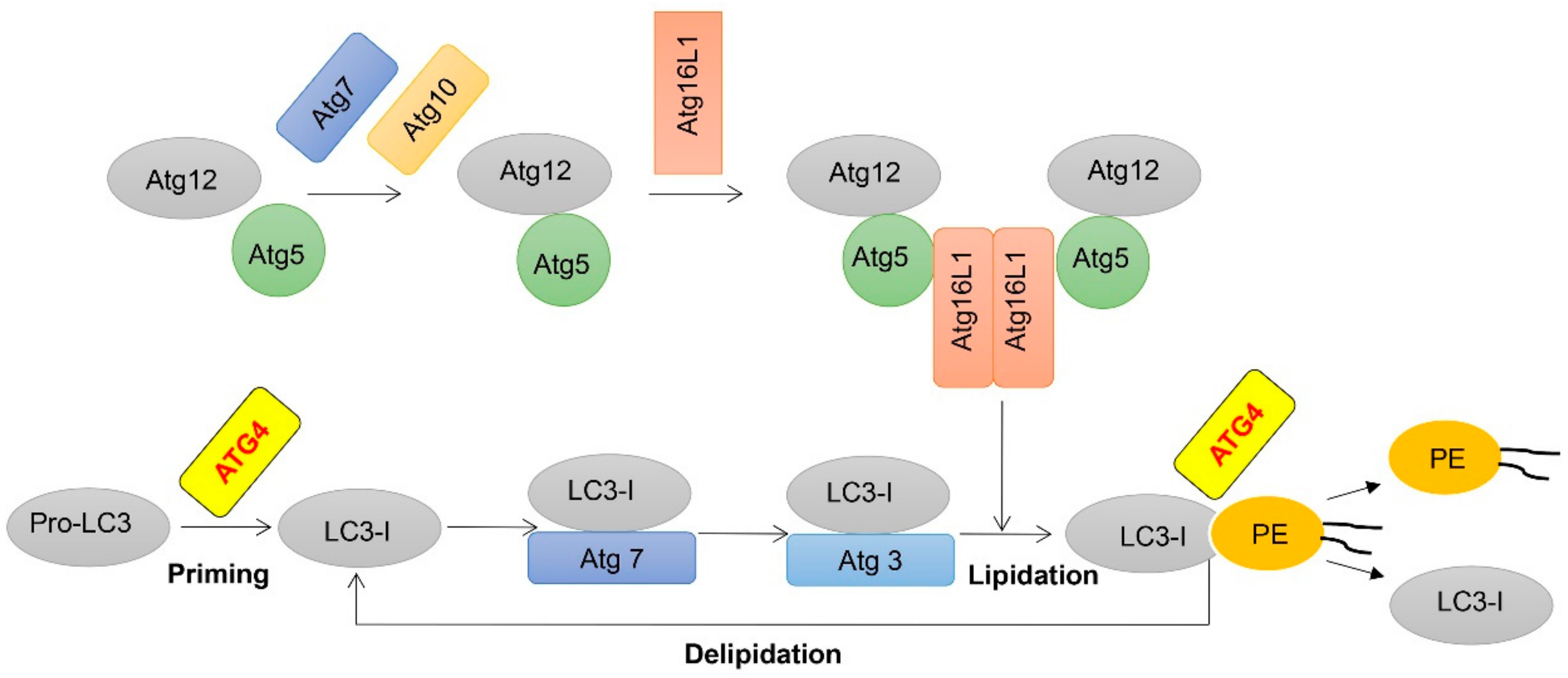
2. A Mechanistic Understanding of ATG4 in Autophagy
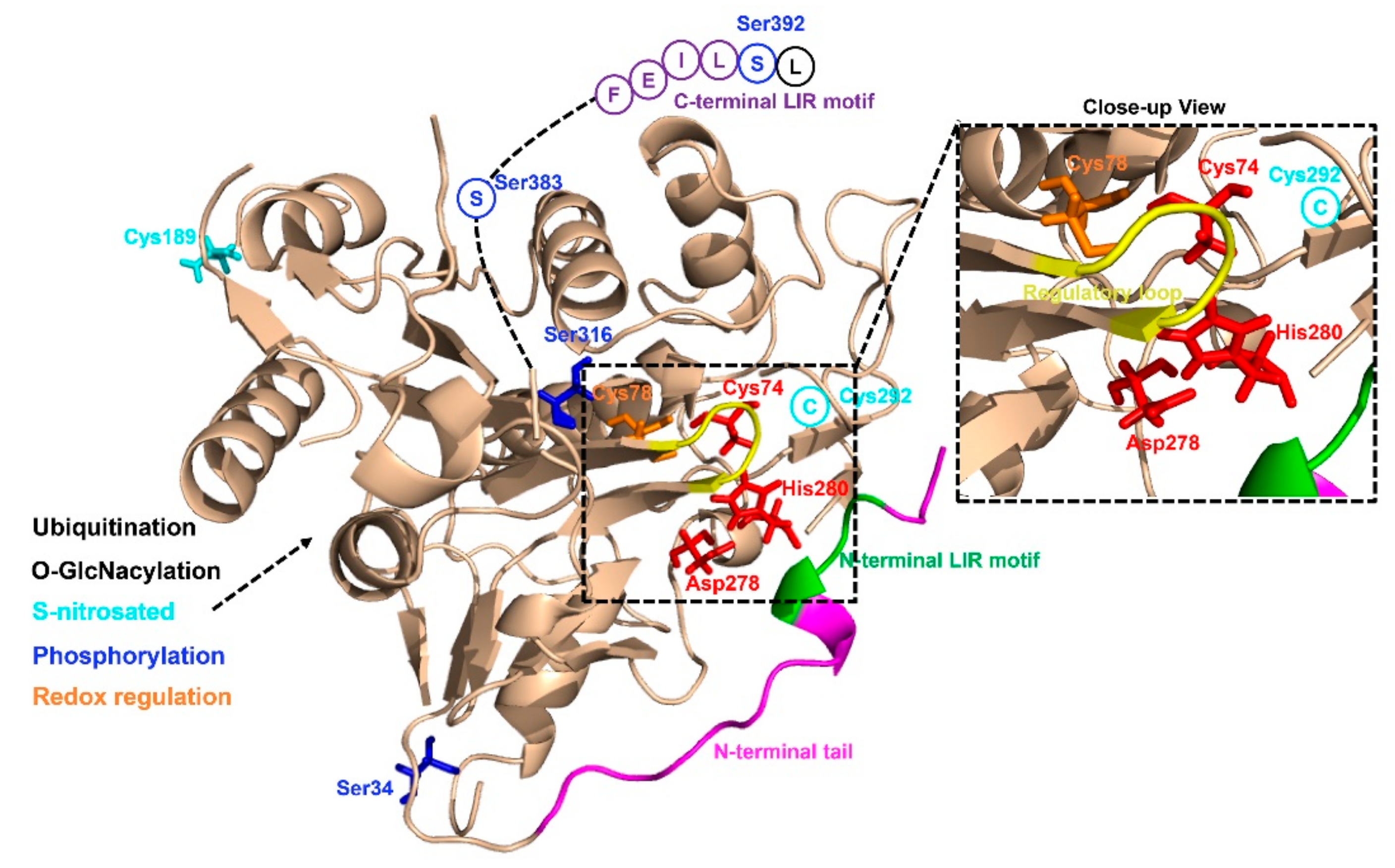
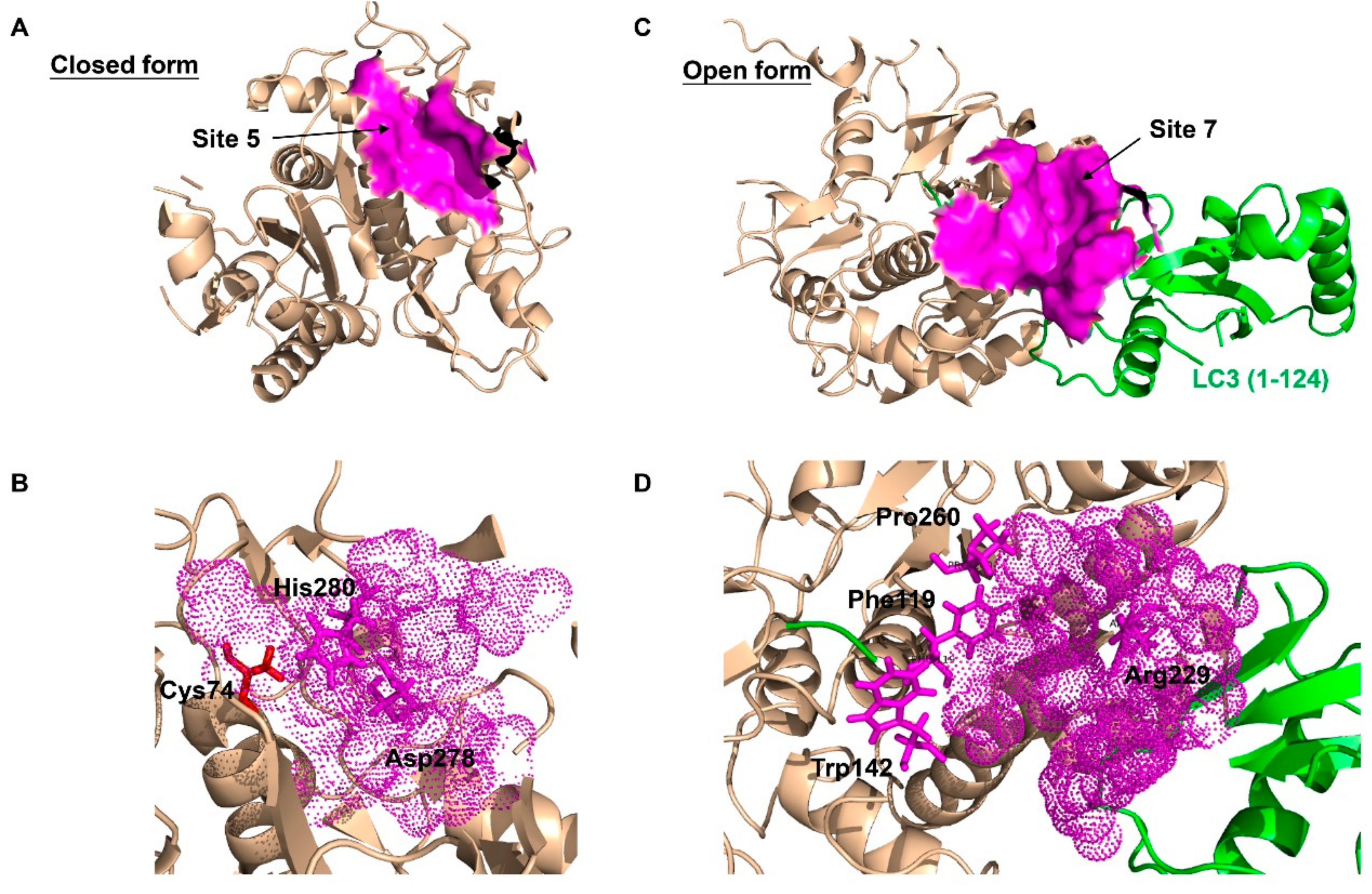
3. The Structure and Post-Translational Modifications of ATG4
4. ATG4 and Cancer
5. Pharmacological Targeting of ATG4B in Cancer Therapy
6. Prospect of the Cancer Therapy Targeting ATG4B Inhibition
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Feng, Y.C.; He, D.; Yao, Z.Y.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Ni, T.; Hong, B.; Wang, H.Y.; Jiang, F.J.; Zou, S.; Chen, Y.; Zheng, X.L.; Klionsky, D.J.; Liang, Y.; et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hou, Y.; Wang, J.; Chen, X.; Shao, Z.-M.; Yin, X.-M. Kinetics Comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 2011, 286, 7327–7338. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Y.; Hong, L.; Xu, J.C.; Zhong, G.P.; Gu, Q.; Gu, Q.Q.; Guan, Y.P.; Zheng, X.P.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ishii, J.; Asai, E.; Ohsumi, Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 2012, 8, 177–186. [Google Scholar] [CrossRef]
- Marino, G.; Fernandez, A.F.; Cabrera, S.; Lundberg, Y.W.; Cabanillas, R.; Rodriguez, F.; Salvador-Montoliu, N.; Vega, J.A.; Germana, A.; Fueyo, A.; et al. Autophagy is essential for mouse sense of balance. J. Clin. Investig. 2010, 120, 2331–2344. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; Lopez-Otin, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/Autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Kuma, A.; Mizushima, N. Transgenic rescue of Atg5-null mice from neonatal lethality with neuron-specific expression of ATG5: Systemic analysis of adult Atg5-deficient mice. Autophagy 2017, 13, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Li, X.F.; Wang, W.X.; Ouedraogo, K.C.; Li, Y.; Gan, C.P.; Tan, S.R.; Zhou, X.K.; Wu, M. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am. J. Physiol. Lung C 2014, 307, L355–L363. [Google Scholar] [CrossRef]
- Bechtel, W.; Helmstadter, M.; Balica, J.; Hartleben, B.; Kiefer, B.; Hrnjic, F.; Schell, C.; Kretz, O.; Liu, S.Y.; Geist, F.; et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J. Am. Soc. Nephrol. 2013, 24, 727–743. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.D.; Zhao, J.J. TFEB Participates in the A beta-induced pathogenesis of Alzheimer’s disease by regulating the autophagy-lysosome pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hong, M.J.; Sun, H.H.; Wang, L.; Shi, X.R.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 2014, 20, 507–514. [Google Scholar] [CrossRef]
- Zhang, L.M.; Ai, Y.H.; Tsung, A. Clinical application: Restoration of immune homeostasis by autophagy as a potential therapeutic target in sepsis. Exp. Ther. Med. 2016, 11, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Danon, M.J.; Takikita, S.; Ralston, E.; Plotz, P.H. The role of autophagy in pathogenesis of Pompe disease. Ann. Neurol. 2007, 62, S64. [Google Scholar]
- Yang, Z.N.J.; Chee, C.E.; Huang, S.B.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Bishop, E.; Bradshaw, T.D. Autophagy modulation: A prudent approach in cancer treatment? Cancer Chemother. Pharm. 2018, 82, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Valcifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J.Y. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef]
- Junco, J.J.; Mancha-Ramirez, A.; Malik, G.; Wei, S.J.; Kim, D.J.; Liang, H.Y.; Slaga, T.J. Ursolic acid and resveratrol synergize with chloroquine to reduce melanoma cell viability. Melanoma Res. 2015, 25, 103–112. [Google Scholar] [CrossRef]
- Xiao, L.; JiaCheng, T.; YueLong, L.; Jin, R.; Cai, X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci. 2014, 4, 10. [Google Scholar]
- Liang, D.H.; El-Zein, R.; Dave, B. Autophagy inhibition to increase radiosensitization in breast cancer. J. Nucl. Med. Radiat. Ther. 2015, 6. [Google Scholar] [CrossRef]
- Qin, L.; Xu, T.Y.; Xia, L.L.; Wang, X.J.; Zhang, X.; Zhang, X.H.; Zhu, Z.W.; Zhong, S.; Wang, C.D.; Shen, Z.J. Chloroquine enhances the efficacy of cisplatin by suppressing autophagy in human adrenocortical carcinoma treatment. Drug Des. Dev. Ther. 2016, 10, 1035–1045. [Google Scholar] [CrossRef]
- Gong, C.; Hu, C.; Gu, F.; Xia, Q.; Yao, C.; Zhang, L.; Qiang, L.; Gao, S.; Gao, Y. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J. Control. Release 2017, 266, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme—A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Eng, C.H.; Wang, Z.C.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.M.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.A.; Wittenburg, L.A.; Amaravadi, R.K.; Gustafson, D.L.; Thorburn, A.; Thamm, D.H. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014, 10, 1415–1425. [Google Scholar] [CrossRef]
- McAfee, Q.; Zhang, Z.H.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.F.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. Iscience 2018, 8, 74–84. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Choutka, C.; Horlings, H.M.; Leung, S.; Baker, J.H.; Lebovitz, C.; Dragowska, W.H.; Go, N.E.; Bally, M.B.; Minchinton, A.I.; et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget 2016, 7, 66970–66988. [Google Scholar] [CrossRef]
- Rothe, K.; Lin, H.; Lin, K.B.L.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Chow, A.; Goda, T.; Wong, A.; Blakely, K.; Rocha, M.; Taeb, S.; Hoang, V.C.; Liu, S.K.; Emmenegger, U. Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochem. Biophys. Res. Commun. 2013, 441, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Wan, Y.Y.; Gong, A.H.; Ge, L.; Jin, J.; Xu, M.; Wu, C.Y. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis 2017, 6, e292. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2003, 22, 4577. [Google Scholar]
- Shintani, T.; Huang, W.P.; Stromhaug, P.E.; Klionsky, D.J. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell 2002, 3, 825–837. [Google Scholar] [CrossRef]
- Li, M.; Fu, Y.; Yang, Z.; Yin, X.M. Measurement of the activity of the Atg4 cysteine proteases. Method Enzymol. 2017, 587, 207–225. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chen, J.J.; Huang, I.F.; Tseng, H.H.; Chang, H.W.; Shu, C.W. Ablation of ATG4B suppressed autophagy and activated AMPK for cell cycle arrest in cancer cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 728–740. [Google Scholar] [CrossRef]
- Liu, P.F.; Tsai, K.L.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chang, H.W.; Shiau, C.W.; Goan, Y.G.; Tseng, H.H.; Wu, C.H.; et al. Drug repurposing screening identifies tioconazole as an ATG4 Inhibitor that suppresses autophagy and sensitizes cancer cells to chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef]
- Wang, W.; Chen, Z.X.; Billiar, T.R.; Stang, M.T.; Gao, W.T. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PLoS ONE 2013, 8, e74222. [Google Scholar] [CrossRef]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.P.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef]
- Sugawara, K.; Suzuki, N.N.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J. Biol. Chem. 2005, 280, 40058–40065. [Google Scholar] [CrossRef]
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618. [Google Scholar] [CrossRef]
- Rasmussen, M.S.; Mouilleron, S.; Shrestha, B.K.; Wirth, M.; Lee, R.; Larsen, K.B.; Princely, Y.A.; O’Reilly, N.; Sjottem, E.; Tooze, S.A.; et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 2017, 13, 834–853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.J.; Ouyang, L.; Liu, B.; Cheng, Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.S.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef]
- Perez-Perez, M.E.; Zaffagnini, M.; Marchand, C.H.; Crespo, J.L.; Lemaire, S.D. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy 2014, 10, 1953–1964. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.K.; Park, N.Y.; Park, S.J.; Kim, B.G.; Shin, J.H.; Jo, D.S.; Bae, D.J.; Suh, Y.A.; Chang, J.H.; Lee, E.K.; et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget 2016, 7, 57186–57196. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Jin, W.H.; Sheng, Q.H.; Shieh, C.H.; Wu, J.R.; Zeng, R. Protein phosphorylation and expression profiling by Yin-yang multidimensional liquid chromatography (Yin-yang MDLC) mass spectrometry. J. Proteome Res. 2007, 6, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Villen, J.; Beausoleil, S.A.; Gerber, S.A.; Gygi, S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA 2007, 104, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Ren, S.B.; Cox, J.; Olsen, J.V.; Macek, B.; Oroshi, M.; Mann, M. PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007, 8, R250. [Google Scholar] [CrossRef]
- Yang, Z.; Wilkie-Grantham, R.P.; Yanagi, T.; Shu, C.W.; Matsuzawa, S.; Reed, J.C. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 2015, 290, 26549–26561. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef]
- Ni, Z.H.; He, J.T.; Wu, Y.R.; Hu, C.J.; Dai, X.F.; Yan, X.J.; Li, B.; Li, X.Z.; Xiong, H.J.; Li, Y.M.; et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy 2018, 14, 685–701. [Google Scholar] [CrossRef]
- Pengo, N.; Agrotis, A.; Prak, K.; Jones, J.; Ketteler, R. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 2017, 8, 294. [Google Scholar] [CrossRef]
- Sanchez-Wandelmer, J.; Reggiori, F. Atg4 in autophagosome biogenesis. Oncotarget 2017, 8, 108290–108291. [Google Scholar] [CrossRef]
- Kuang, E.; Okumura, C.Y.M.; Sheffy-Levin, S.; Varsano, T.; Shu, V.C.W.; Qi, J.F.; Niesman, I.R.; Yang, H.J.; Lopez-Otin, C.; Yang, W.Y.; et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Ozpolat, B.; Benbrook, D.M. Targeting autophagy in cancer management—Strategies and developments. Cancer Manag. Res. 2015, 7, 291–299. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.H.; Mukherjee, C.; Shi, Y.F.; Gelinas, C.; Fan, Y.J.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Luo, T.; Fu, J.; Xu, A.; Su, B.; Ren, Y.B.; Li, N.; Zhu, J.J.; Zhao, X.F.; Dai, R.Y.; Cao, J.; et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 2016, 12, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Jiang, L.L.; Fu, X.; Wang, W.J.; Ma, J.Q.; Tian, T.; Nan, K.J.; Liang, X. Cytoplasmic liver kinase B1 promotes the growth of human lung adenocarcinoma by enhancing autophagy. Cancer Sci. 2018, 109, 3055–3067. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Wei, H.J.; Wei, S.; Gan, B.Y.; Peng, X.; Zou, W.P.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Gene Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Yang, S.H.; Wang, X.X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.Q.; Bause, A.; Li, Y.H.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Gene Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Liu, P.F.; Leung, C.M.; Chang, Y.H.; Cheng, J.S.; Chen, J.J.; Weng, C.J.; Tsai, K.W.; Hsu, C.J.; Liu, Y.C.; Hsu, P.C.; et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy 2014, 10, 1454–1465. [Google Scholar] [CrossRef]
- Toshima, T.; Shirabe, K.; Matsumoto, Y.; Yoshiya, S.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Maehara, Y. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial beta-oxidation. J. Gastroenterol. 2014, 49, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Su, J.; Qian, H.; Guo, T. SLC27A4 regulate ATG4B activity and control reactions to chemotherapeutics-induced autophagy in human lung cancer cells. Tumor Biol. 2016, 37, 6943–6952. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Meyer, L.; Chang, D.W.; Lin, J.; Pu, X.; Ye, Y.Q.; Gu, J.A.; Wu, X.F.; Lu, K. Genetic variants in MicroRNA biosynthesis pathways and binding sites modify ovarian cancer risk, survival, and treatment response. Cancer Res. 2010, 70, 9765–9776. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.P.; Chen, L.Y.; Huang, R.L.; Su, P.H.; Chan, M.W.Y.; Chang, C.C.; Yu, M.H.; Wang, P.H.; Yen, M.S.; Nephew, K.P.; et al. Hypomethylation signature of tumor-initiating cells predicts poor prognosis of ovarian cancer patients. Hum. Mol. Genet. 2014, 23, 1894–1906. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.J.; Wu, L.X.; Wang, W.; Ye, Y.Y.; Yang, J.; Chen, H.; Yang, Q.F.; Zhang, X.Y.; Wang, B.; Chen, W.X. Nucleotide variation in ATG4A and susceptibility to cervical cancer in Southwestern Chinese women. Oncol. Lett. 2018, 15, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Lu, Y.Y.; Hu, S.L.; Huang, Q.; Li, S.J.; Huang, Y.; Hu, Q.; Wu, L.X.; Chen, W.X. An intron SNP rs807185 in ATG4A decreases the risk of lung cancer in a southwest Chinese population. Eur. J. Cancer Prev. 2016, 25, 255–258. [Google Scholar] [CrossRef]
- Antonelli, M.; Strappazzon, F.; Arisi, I.; Brandi, R.; D’Onofrio, M.; Sambucci, M.; Manic, G.; Vitale, I.; Barila, D.; Stagni, V. ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 2017, 8, 21692–21709. [Google Scholar] [CrossRef]
- Frankel, L.B.; Wen, J.; Lees, M.; Hoyer-Hansen, M.; Farkas, T.; Krogh, A.; Jaattela, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. Embo J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef]
- Xu, Y.H.; An, Y.; Wang, Y.; Zhang, C.H.; Zhang, H.; Huang, C.J.; Jiang, H.; Wang, X.H.; Li, X.C. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013, 29, 2019–2024. [Google Scholar] [CrossRef]
- Gil, J.; Ramsey, D.; Pawlowski, P.; Szmida, E.; Leszczynski, P.; Bebenek, M.; Sasiadek, M.M. The influence of tumor microenvironment on ATG4D gene expression in colorectal cancer patients. Med. Oncol. 2018, 35, 159. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Chen, H.Y.; White, E. Role of autophagy in cancer prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef]
- Wu, Z.J.; Chang, P.C.; Yang, J.C.; Chu, C.Y.; Wang, L.Y.; Chen, N.T.; Ma, A.H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Cancer Res. 2010, 70, AM10–AM4684. [Google Scholar] [CrossRef]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Enzenmuller, S.; Gonzalez, P.; Debatin, K.M.; Fulda, S. Chloroquine overcomes resistance of lung carcinoma cells to the dual PI3K/mTOR inhibitor PI103 by lysosome-mediated apoptosis. Anti Cancer Drugs 2013, 24, 14–19. [Google Scholar] [CrossRef]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, R.; Seed, B. Quantitation of autophagy by luciferase release assay. Autophagy 2008, 4, 801–806. [Google Scholar] [CrossRef][Green Version]
- Shu, C.W.; Liu, P.F.; Huang, C.M. High throughput screening for drug discovery of autophagy modulators. Comb. Chem. High Throughput Screen. 2012, 15, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.; Ye, Q.Z.; Vogt, A.; Yin, X.M. A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy 2012, 8, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Q.; Xu, Z.H.; Han, L.; Liu, C.; Zhou, Z.; Qiu, Z.X.; Lin, X.F.; Tang, G.Z.; Shen, H.; Aebi, J.; et al. Identification of new ATG4B inhibitors based on a novel high-throughput screening platform. Slas Discov. 2017, 22, 338–347. [Google Scholar] [CrossRef][Green Version]
- Qiu, Z.X.; Kuhn, B.; Aebi, J.; Lin, X.F.; Ding, H.Y.; Zhou, Z.; Xu, Z.H.; Xu, D.Q.; Han, L.; Liu, C.; et al. Discovery of fluoromethylketone-based peptidomimetics as covalent ATG4B (Autophagin-1) inhibitors. ACS Med. Chem. Lett. 2016, 7, 802–806. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Bosc, D.; Vezenkov, L.; Bortnik, S.; An, J.; Xu, J.; Choutka, C.; Hannigan, A.M.; Kovacic, S.; Loo, S.; Clark, P.G.K.; et al. A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease ATG4B. Sci. Rep. 2018, 8, 11653. [Google Scholar] [CrossRef] [PubMed]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef]
- Choi, K.-M.; Nam, H.Y.; Na, J.H.; Kim, S.W.; Kim, S.Y.; Kim, K.; Kwon, I.C.; Ahn, H.J. A monitoring method for Atg4 activation in living cells using peptide-conjugated polymeric nanoparticles. Autophagy 2011, 7, 1052–1062. [Google Scholar] [CrossRef][Green Version]
- Ni, Z.; Gong, Y.; Dai, X.; Ding, W.; Wang, B.; Gong, H.; Qin, L.; Cheng, P.; Li, S.; Lian, J.; et al. AU4S: A novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy 2015, 11, 403–415. [Google Scholar] [CrossRef][Green Version]
- Shu, C.W.; Madiraju, C.; Zhai, D.; Welsh, K.; Diaz, P.; Sergienko, E.; Sano, R.; Reed, J.C. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J. Biomol. Screen. 2011, 16, 174–182. [Google Scholar] [CrossRef]
- Chu, J.; Fu, Y.; Xu, J.; Zheng, X.; Gu, Q.; Luo, X.; Dai, Q.; Zhang, S.; Liu, P.; Hong, L.; et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch. Biochem. Biophys. 2018, 644, 29–36. [Google Scholar] [CrossRef]
- Vezenkov, L.; Honson, N.S.; Kumar, N.S.; Bosc, D.; Kovacic, S.; Nguyen, T.G.; Pfeifer, T.A.; Young, R.N. Development of fluorescent peptide substrates and assays for the key autophagy-initiating cysteine protease enzyme, ATG4B. Bioorg. Med. Chem. 2015, 23, 3237–3247. [Google Scholar] [CrossRef]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef]
- Marino, G.; Uria, J.A.; Puente, X.S.; Quesada, V.; Bordallo, J.; Lopez-Otin, C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J. Biol. Chem. 2003, 278, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; MacVicar, T.D.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy 2012, 8, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.S.; Lane, J.D. Atg4D at the interface between autophagy and apoptosis. Autophagy 2009, 5, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Gorski, S.M. Clinical applications of autophagy proteins in cancer: From potential targets to biomarkers. Int. J. Mol. Sci. 2017, 18, 1496. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
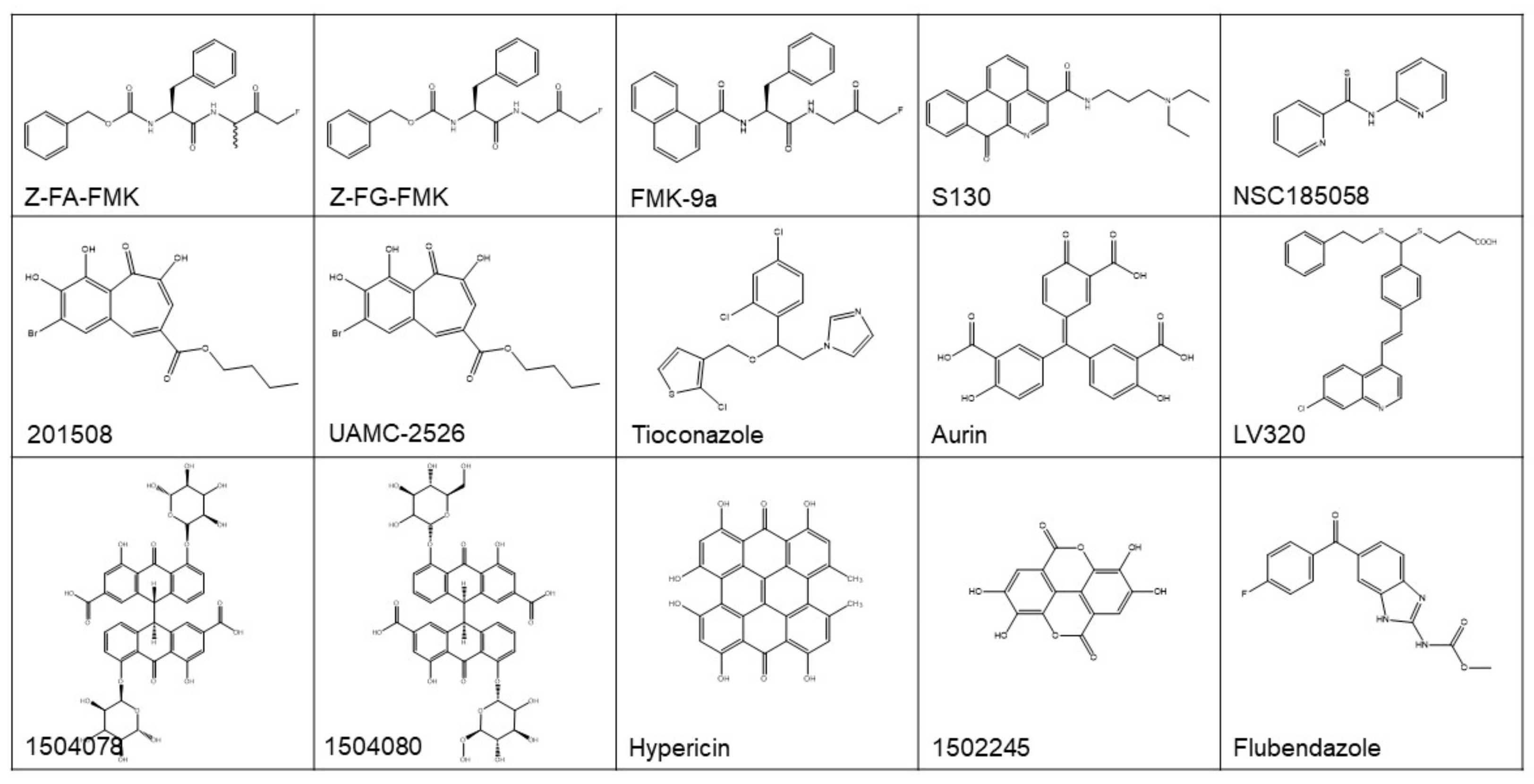
| Name/Compound ID | IC50 | Methods | Effect on ATG4B | Effect on Autophagy | Effect on Cancer | Ref. |
|---|---|---|---|---|---|---|
| 201508 | 2.3 µM | LC3B-PLA2 assay | Inhibiting | N.R. | N.R. | [107] |
| 1502245 | 3.0 µM | LC3B-PLA2 assay | Inhibiting | N.R. | N.R. | [107] |
| 1504078 | 1.7 µM | LC3B-PLA2 assay | Inhibiting | N.R. | N.R. | [107] |
| 1504080 | 1.1 µM | LC3B-PLA2 assay | Inhibiting | N.R. | N.R. | [107] |
| Tioconazole | 1.8 µM | LC3B-PLA2 assay | Inhibiting | Inhibiting | Enhancing Dox-induced cytotoxicity in colorectal cancer | [44] |
| Z-FA-FMK | 14.8 µM | TR-FRET assay | Inhibiting | N.R. | N.R. | [100] |
| Z-FG-FMK | 1.13 µM | TR-FRET assay | Inhibiting | N.R. | N.R. | [100] |
| FMK-9a | 80 nM | TR-FRET assay | Inhibiting | Inducing | Having no effect on the survival of HeLa cells | [101,108] |
| Hypericin | 30 µM | FRET assay | Inhibiting | N.R. | N.R. | [109] |
| Aurin | 8.8 µM | FRET assay | Inhibiting | N.R. | N.R. | [109] |
| LV-320 | 24.5 µM | Fluorescent peptide substrate assay | Inhibiting | Inhibiting | N.R. | [103] |
| UAMC-2526 | N.R. | LC3-GST cleavage assay | Inhibiting | Inhibiting | Enhancing Oxaliplatin-induced cytotoxicity in colorectal cancer | [110] |
| NSC185058 | 51 µM | In silico screening, LC3-GST cleavage assay | Inhibiting | Inhibiting | Suppressing the development ofSao-2 cells; enhancing the anti-glioblastoma activity of radiation therapy | [31,62] |
| S130 | 3.2 µM | In silico screening, FRET assay | Inhibiting | Inhibiting | Arresting the growth of colorectal cancer | [6] |
| Flubendazole | N.R. | In silico analysis | Inducing | Inducing | Inducing autophagic cell death in MDA-MB-231 cells | [102] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Huang, Z.; Hong, L.; Lu, J.-H.; Feng, D.; Yin, X.-M.; Li, M. Targeting ATG4 in Cancer Therapy. Cancers 2019, 11, 649. https://doi.org/10.3390/cancers11050649
Fu Y, Huang Z, Hong L, Lu J-H, Feng D, Yin X-M, Li M. Targeting ATG4 in Cancer Therapy. Cancers. 2019; 11(5):649. https://doi.org/10.3390/cancers11050649
Chicago/Turabian StyleFu, Yuanyuan, Zhiying Huang, Liang Hong, Jia-Hong Lu, Du Feng, Xiao-Ming Yin, and Min Li. 2019. "Targeting ATG4 in Cancer Therapy" Cancers 11, no. 5: 649. https://doi.org/10.3390/cancers11050649
APA StyleFu, Y., Huang, Z., Hong, L., Lu, J.-H., Feng, D., Yin, X.-M., & Li, M. (2019). Targeting ATG4 in Cancer Therapy. Cancers, 11(5), 649. https://doi.org/10.3390/cancers11050649