A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth


 ,
,
Abstract
1. Introduction
2. Results
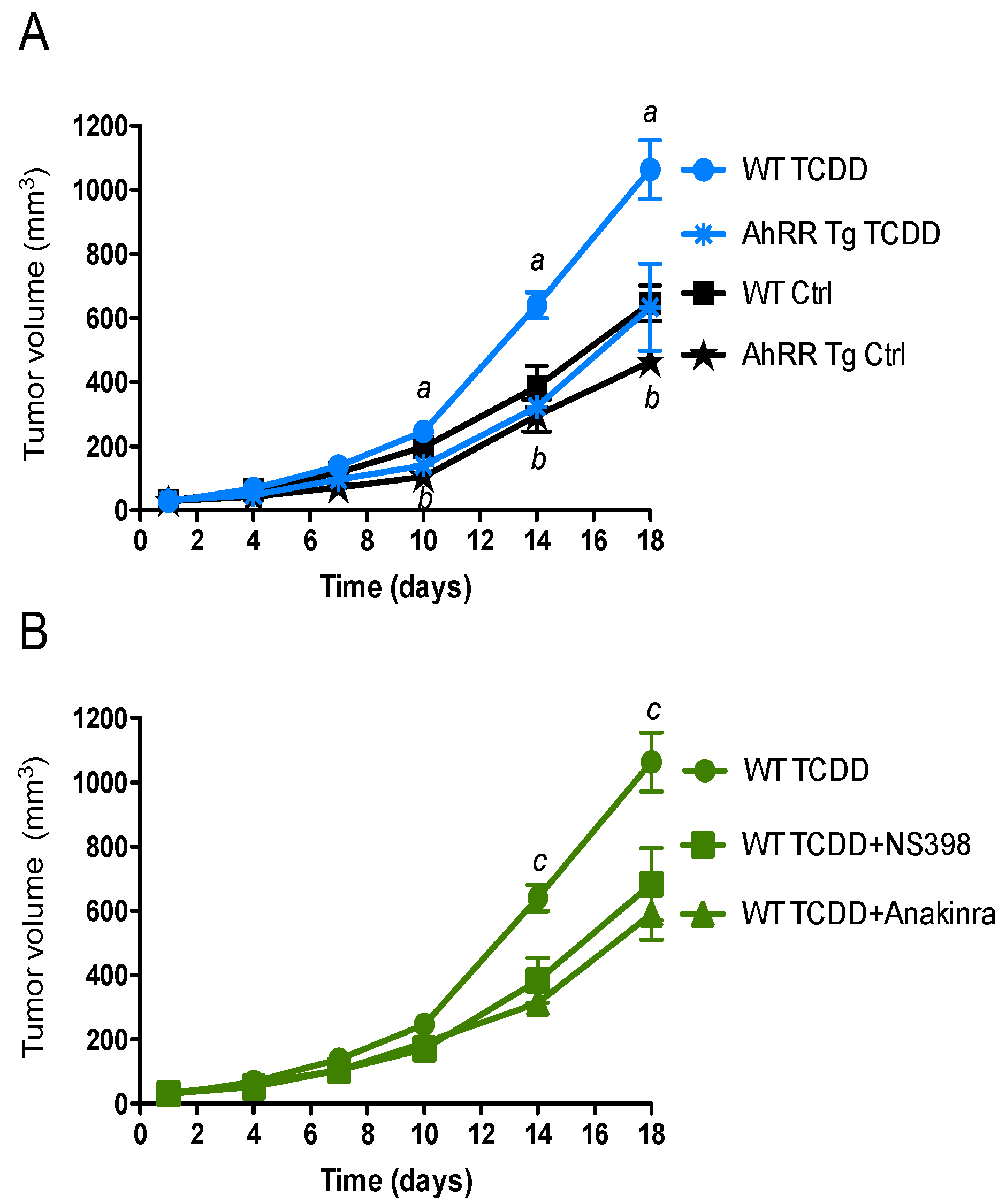
2.1. Suppression of Tumor Growth in AhRR Tg Mice
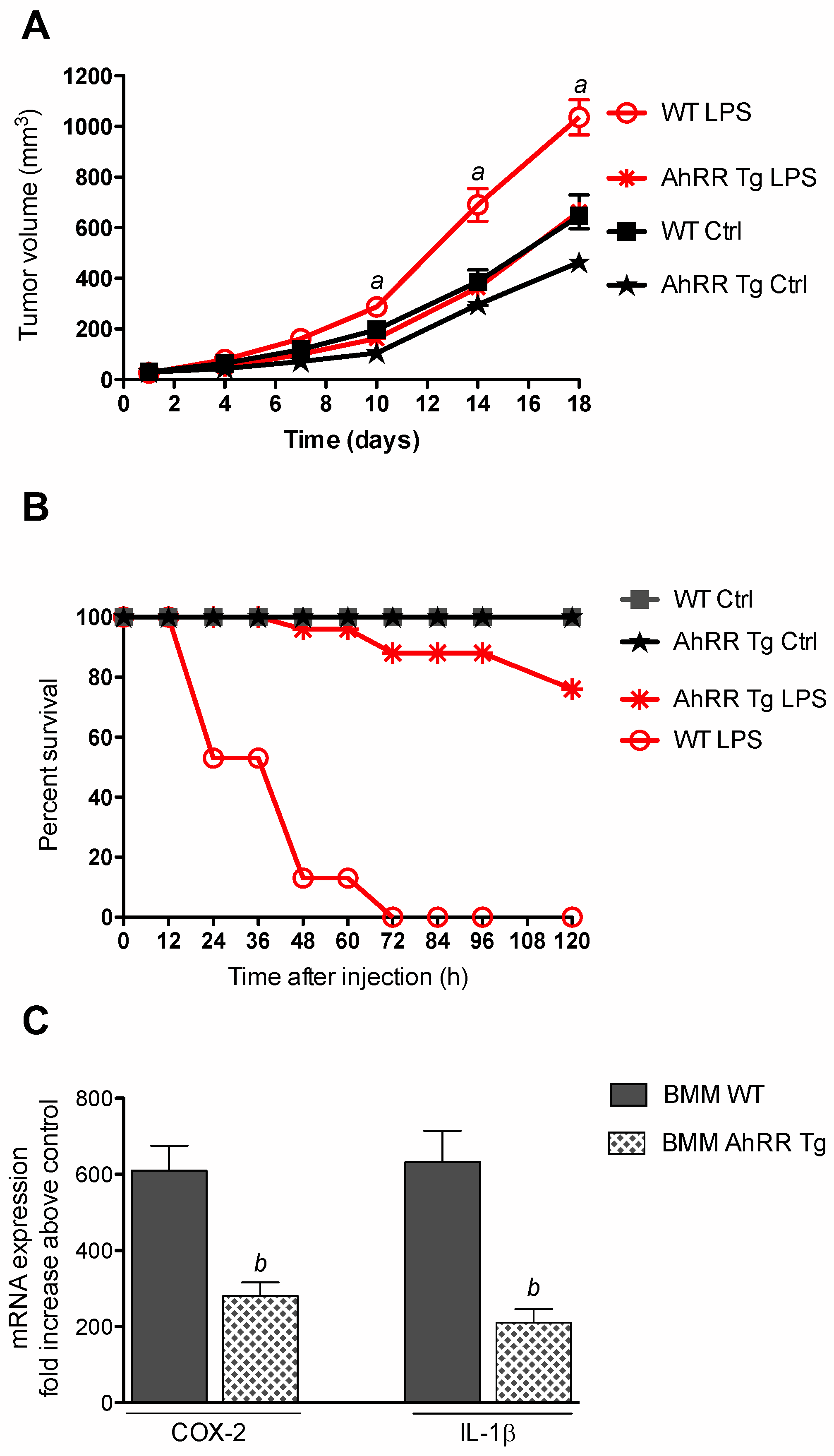
2.2. Resistance of AhRR Tg Mice to LPS
2.3. AhRR Suppresses TCDD-Induced Expression of Inflammatory Markers
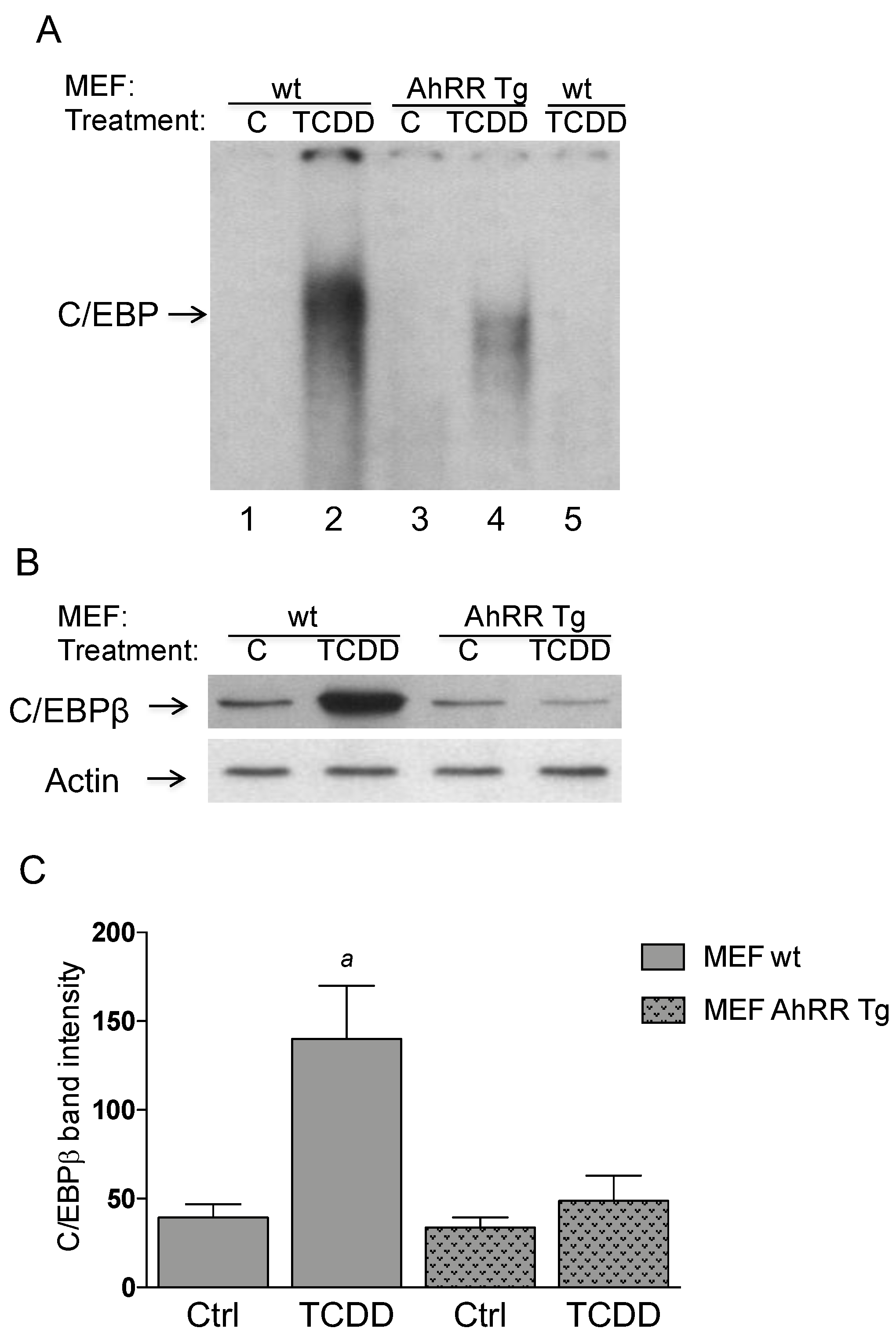
2.4. Repressed DNA-Binding Activity of C/EBP in AhRR Tg MEF
2.5. TCDD-Induced PKA Activity is Reduced in AhRR Tg MEF
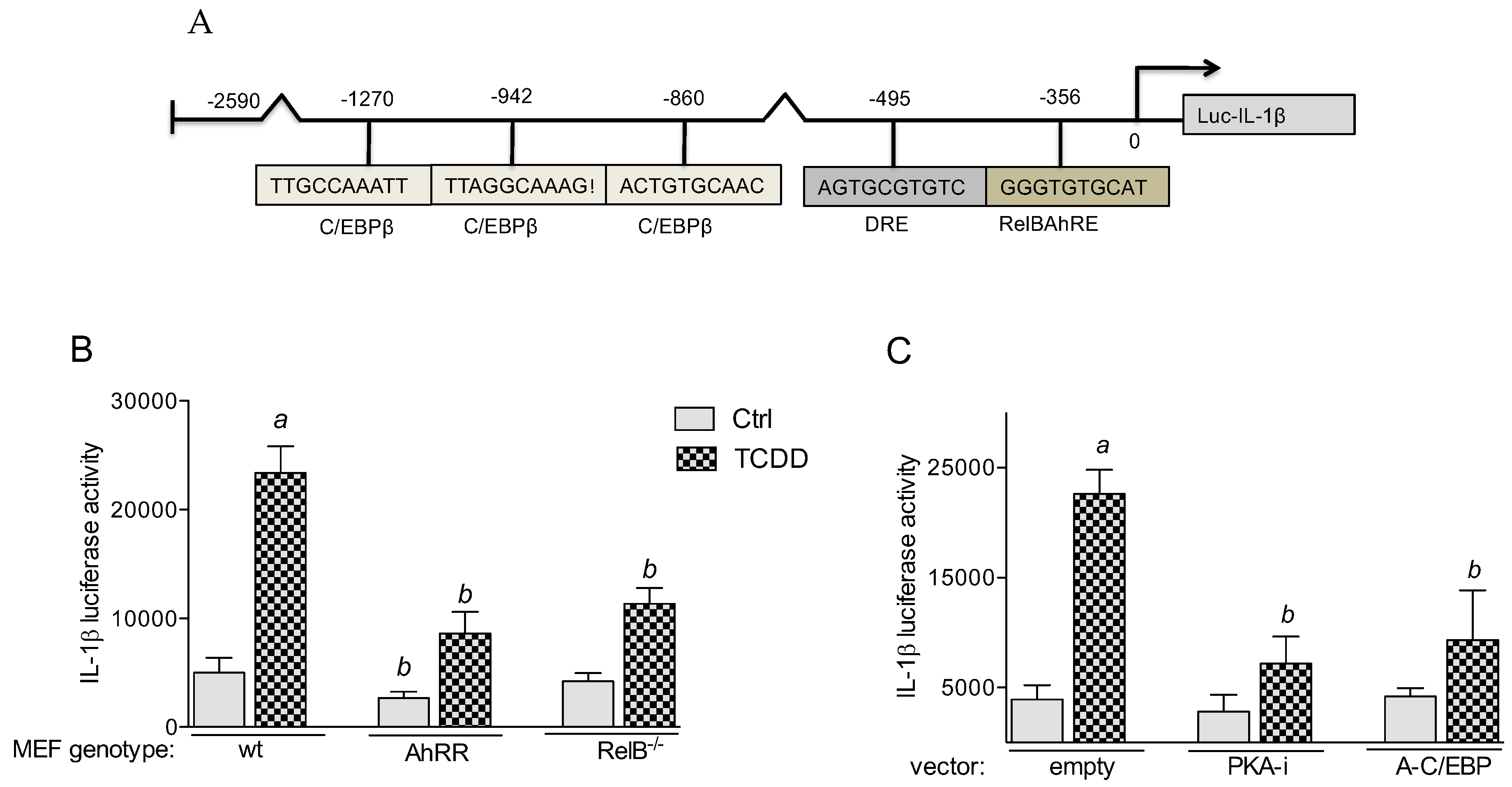
2.5.1. PKA and C/EBPβ Mediate TCDD-Induced IL-1β Promoter Activity
2.5.2. Enhanced Recruitment of AhR to a RelBAhRE Binding Site of the IL-1β Promoter
2.5.3. Physical Association of AhRR, ARNT, and RelB
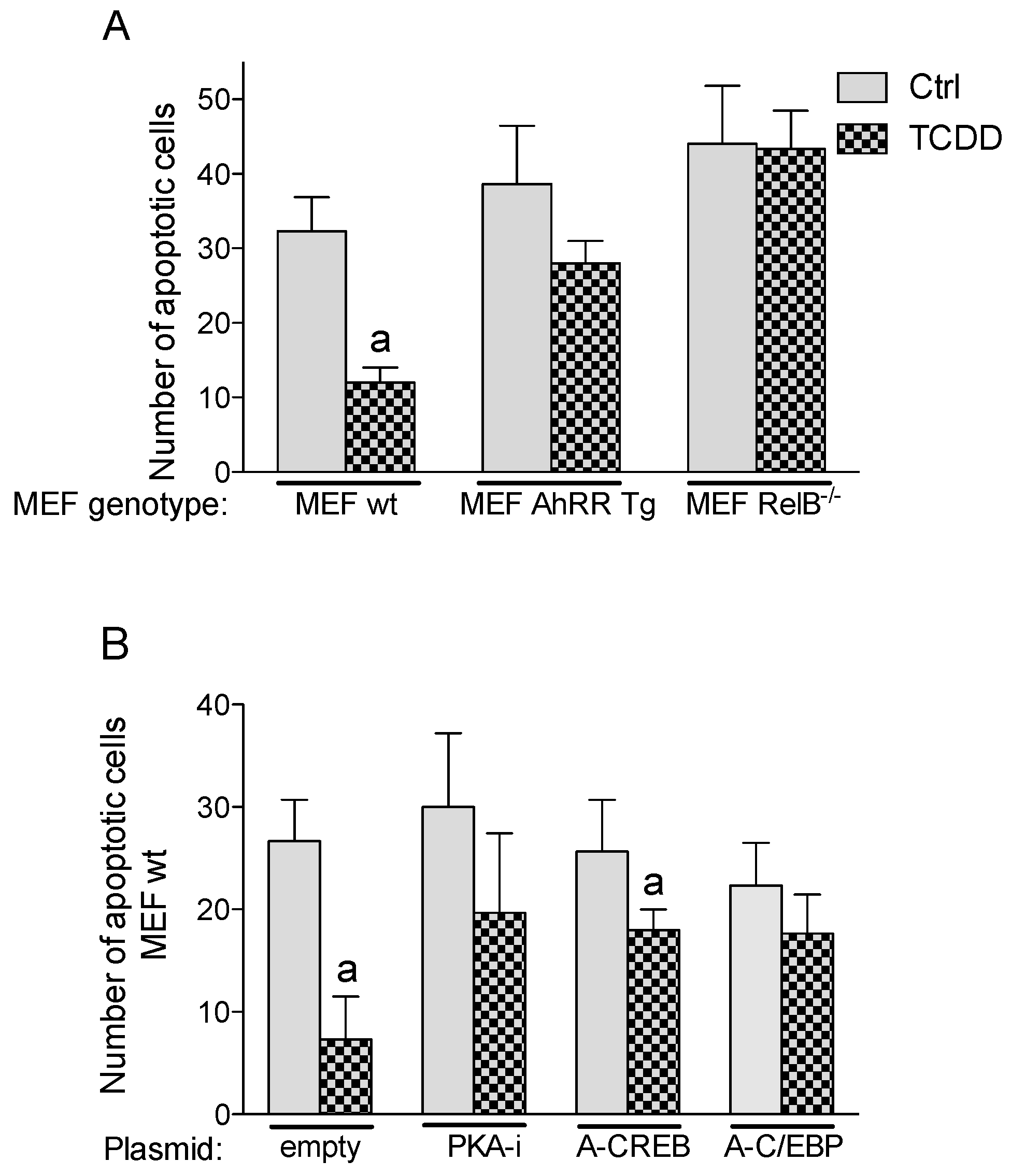
2.6. Inhibition of TCDD-Induced Apoptotic Resistance by AhRR
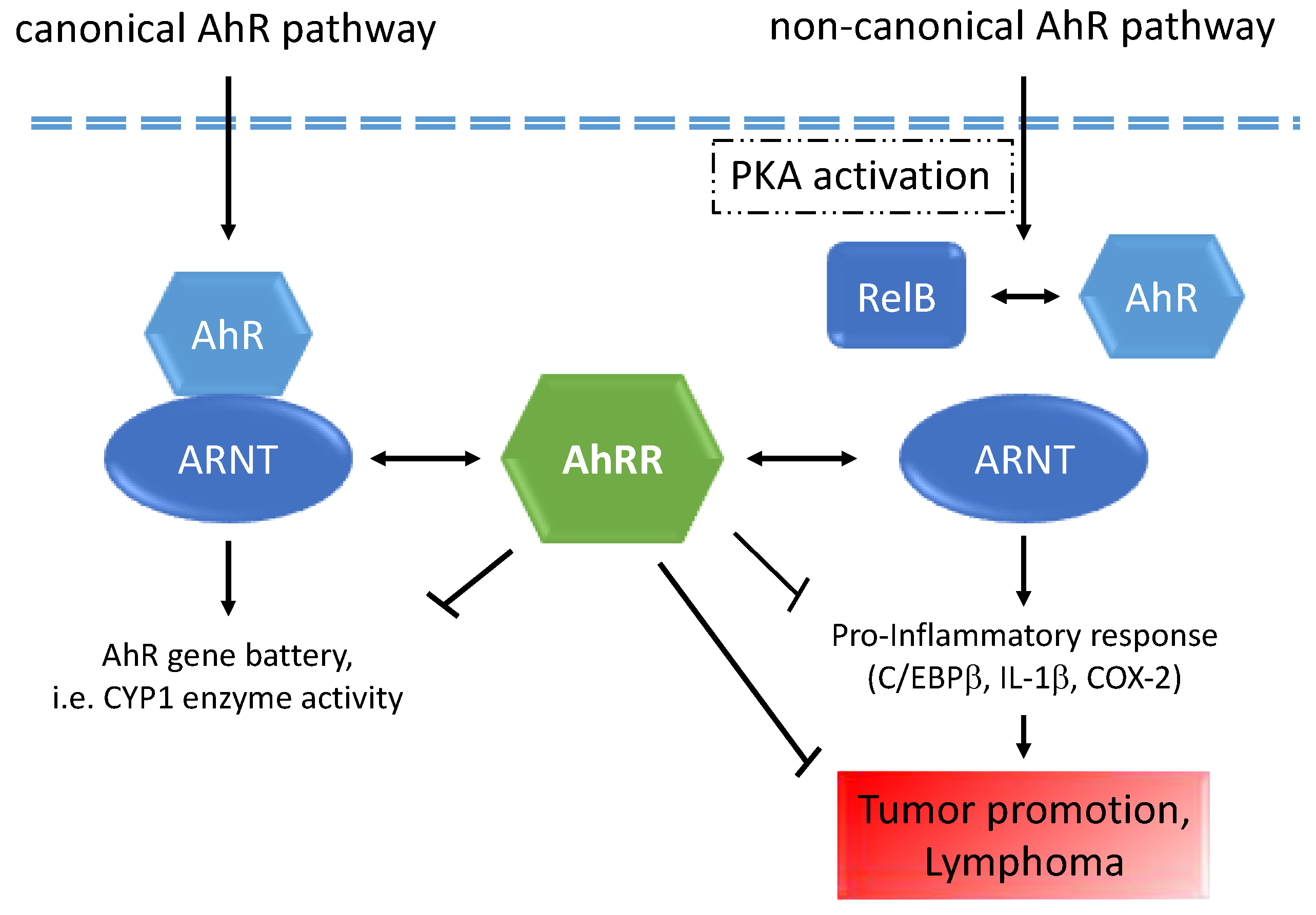
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Transfection Experiments
4.3. Mice and Treatment
4.4. Electrophoretic Mobility Shift Assay (EMSA)
4.5. RNA Isolation and Real-Time PCR
4.6. Protein Kinase A (PKA) Assays
4.7. ChIP Assay
4.8. Nuclear Complex Co-Immunoprecipitation Assay
4.9. Western Blot Analysis
4.10. Apoptosis Assay on UV-Irradiated Cells
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van den Berg, M.; Denison, M.S.; Birnbaum, L.S.; Devito, M.J.; Fiedler, H.; Falandysz, J.; Rose, M.; Schrenk, D.; Safe, S.; Tohyama, C.; et al. Polybrominated dibenzo-p-dioxins, dibenzofurans, and biphenyls: Inclusion in the toxicity equivalency factor concept for dioxin-like compounds. Toxicol. Sci. 2013, 133, 197–208. [Google Scholar] [CrossRef]
- Pitot, H.C.; Goldsworthy, T.; Campbell, H.A.; Poland, A. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Res. 1980, 40, 3616–3620. [Google Scholar] [PubMed]
- Castro, D.J.; Löhr, C.V.; Fischer, K.A.; Pereira, C.B.; Williams, D.E. Lymphoma and lung cancer in offspring born to pregnant mice dosed with dibenzo[a,l]pyrene: The importance of in utero vs. lactational exposure. Toxicol. Appl. Pharmacol. 2008, 233, 454–468. [Google Scholar] [CrossRef]
- Vogel, C.F.; Li, W.; Sciullo, E.; Newman, J.; Hammock, B.; Reader, J.R.; Tuscano, J.; Matsumura, F. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am. J. Pathol. 2007, 171, 1538–1548. [Google Scholar] [CrossRef]
- Landgren, O.; Shim, Y.K.; Michalek, J.; Costello, R.; Burton, D.; Ketchum, N.; Calvo, K.R.; Caporaso, N.; Raveche, E.; Middleton, D.; et al. Agent orange exposure and monoclonal gammopathy of undetermined significance: An operation ranch hand veteran cohort study. JAMA Oncol. 2015, 1, 1061–1068. [Google Scholar]
- Xu, J.; Ye, Y.; Huang, F.; Chen, H.; Wu, H.; Huang, J.; Hu, J.; Xia, D.; Wu, Y. Association between dioxin and cancer incidence and mortality: A meta-analysis. Sci. Rep. 2016, 6, 38012. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Dragan, Y.P.; Schrenk, D. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Addit. Contam. 2000, 4, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of constitutive and inducible AHR signaling: Complex interactions involving the AHR repressor. Biochem. Pharmacol. 2009, 77, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor repressor—More than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer. Curr. Opin. Toxicol. 2017, 2, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Tigges, J.; Weighardt, H.; Wolff, S.; Götz, C.; Förster, I.; Kohne, Z.; Huebenthal, U.; Merk, H.F.; Abel, J.; Haarmann-Stemmann, T.; et al. Aryl hydrocarbon receptor repressor (AhRR) function revisited: Repression of CYP1 activity in human skin fibroblasts is not related to AhRR expression. J. Invest. Dermatol. 2013, 133, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Shimizu, T.; Ohto, U. The crystal structure of the AhRR/ARNT heterodimer reveals the structural basis of the repression of AhR-mediated transcription. J. Biol. Chem. 2017, 292, 17609–17616. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.R.; Karchner, S.I.; Allan, L.L.; Pollenz, R.S.; Tanguay, R.L.; Jenny, M.J.; Sherr, D.H.; Hahn, M.E. Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Mol. Pharmacol. 2008, 73, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Chang, W.L.; Kado, S.; McCulloh, K.; Vogel, H.; Wu, D.; Haarmann-Stemmann, T.; Yang, G.; Leung, P.S.; Matsumura, F.; et al. Transgenic overexpression of aryl hydrocarbon receptor repressor (AhRR) and AhR-mediated induction of CYP1A1, cytokines, and acute toxicity. Environ. Health Perspect. 2016, 124, 1071–1083. [Google Scholar] [CrossRef]
- Zudaire, E.; Cuesta, N.; Murty, V.; Woodson, K.; Adams, L.; Gonzalez, N.; Martínez, A.; Narayan, G.; Kirsch, I.; Franklin, W.; et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J. Clin. Invest. 2008, 118, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Schlezinger, J.J.; Liu, D.; Farago, M.; Seldin, D.C.; Belguise, K.; Sonenshein, G.E.; Sherr, D.H. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol. Chem. 2006, 387, 1175–1187. [Google Scholar] [CrossRef]
- Yang, S.Y.; Ahmed, S.; Satheesh, S.V.; Matthews, J. Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells. Arch. Toxicol. 2018, 1, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Richmond, R.; Hu, P.; French, L.; Shin, J.; Bourdon, C. Prenatal exposure to maternal cigarette smoking and DNA methylation: Epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ. Health Perspect. 2015, 123, 193–199. [Google Scholar] [CrossRef]
- Vogel, C.; Boerboom, A.M.; Baechle, C.; El-Bahay, C.; Kahl, R.; Degen, G.H.; Abel, J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: Possible c-Src-mediated pathway. Carcinogenesis 2000, 21, 2267–2274. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Park, S.; Liedtke, C.; Trautwein, C.; Matsumura, F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J. Biol. Chem. 2004, 279, 8886–8894. [Google Scholar] [CrossRef]
- Sutter, T.R.; Guzman, K.; Dold, K.M.; Greenlee, W.F. Targets for dioxin: Genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science 1991, 254, 415–418. [Google Scholar] [CrossRef]
- Brandstätter, O.; Schanz, O.; Vorac, J.; König, J.; Mori, T.; Maruyama, T.; Korkowski, M.; Haarmann-Stemmann, T.; von Smolinski, D.; Schultze, J.L.; et al. Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor. Sci. Rep. 2016, 17, 26091. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 12, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.W.; Duckett, C.S. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-kappaB-dependent transcription. Science 2009, 323, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Gardella, K.A.; Muro, I.; Fang, G.; Sarkar, K.; Mendez, O.; Wright, C.W. Aryl hydrocarbon receptor nuclear translocator (ARNT) isoforms control lymphoid cancer cell proliferation through differentially regulating tumor suppressor p53 activity. Oncotarget 2016, 7, 10710–10722. [Google Scholar] [CrossRef]
- Vogel, C.F.; Li, W.; Wu, D.; Miller, J.K.; Sweeney, C.; Lazennec, G.; Fujisawa, Y.; Matsumura, F. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch. Biochem. Biophys. 2011, 512, 78–86. [Google Scholar] [CrossRef]
- Secchiero, P.; Barbarotto, E.; Gonelli, A.; Tiribelli, M.; Zerbinati, C.; Celeghini, C.; Agostinelli, C.; Pileri, S.A.; Zauli, G. Potential pathogenetic implications of cyclooxygenase-2 overexpression in B chronic lymphoid leukemia cells. Am. J. Pathol. 2005, 167, 1599–1607. [Google Scholar] [CrossRef]
- Wun, T.; McKnight, H.; Tuscano, J.M. Increased cyclooxygenase-2 (COX-2): A potential role in the pathogenesis of lymphoma. Leuk. Res. 2004, 28, 179–190. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Buchanan, F.G.; Holla, V.; Katkuri, S.; Matta, P.; DuBois, R.N. Targeting cyclooxygenase-2 and the epidermal growth factor receptor for the prevention and treatment of intestinal cancer. Cancer Res. 2007, 67, 9380–9388. [Google Scholar] [CrossRef] [PubMed]
- Degner, S.C.; Papoutsis, A.J.; Selmin, O.; Romagnolo, D.F. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3'-diindolylmethane in breast cancer cells. J. Nutr. 2009, 139, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Pande, K.; Moran, S.M.; Bradfield, C.A. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol. Pharmacol. 2005, 67, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.D.; Nukaya, M.; Moran, S.M.; Glover, E.; Weinberg, S.; Balbo, S.; Hecht, S.S.; Pitot, H.C.; Drinkwater, N.R.; Bradfield, C.A. Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors. Toxicol. Sci. 2014, 140, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kaczanowska, S.; Davila, E. Il-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance. Front Immunol. 2014, 5, 553. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Khatami, M.; Baglole, C.J.; Sun, J.; Harris, S.; Moon, E.Y. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 2015, 36, 232–253. [Google Scholar] [CrossRef]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007, 67, 1062–1071. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 9990–9995. [Google Scholar] [CrossRef]
- Basak, C.; Pathak, S.K.; Bhattacharyya, A.; Mandal, D.; Pathak, S.; Kundu, M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and pak1-mediated caspase-1 activation play essential roles in interleukin-1beta release from helicobacter pylori lipopolysaccharide-stimulated macrophages. J. Biol. Chem. 2005, 280, 4279–4288. [Google Scholar] [CrossRef]
- Fletcher, B.S.; Kujubu, D.A.; Perrin, D.M.; Herschman, H.R. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J. Biol. Chem. 1992, 267, 4338–4344. [Google Scholar]
- Oesch-Bartlomowicz, B.; Huelster, A.; Wiss, O.; Antoniou-Lipfert, P.; Dietrich, C.; Arand, M.; Weiss, C.; Bockamp, E.; Oesch, F. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: Divergent signaling pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 9218–9223. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Davis-Turak, J.; Macal, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, E.I.; et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat. Immunol. 2012, 13, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Niehof, M.; Streetz, K.; Rakemann, T.; Bischoff, S.C.; Manns, M.P.; Horn, F.; Trautwein, C. Interleukin-6-induced tethering of STAT3 to the LAP/C/EBP beta promoter suggests a new mechanism of transcriptional regulation by STAT3. J. Biol. Chem. 2001, 276, 9016–9027. [Google Scholar] [CrossRef] [PubMed]
- Wessells, J.; Yakar, S.; Johnson, P.F. Critical prosurvival roles for C/EBP beta and insulin-like growth factor I in macrophage tumor cells. Mol. Cell Biol. 2004, 24, 3238–3250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bundy, L.M.; Sealy, L. CCAAT/enhancer binding protein beta (C/EBPbeta)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene 2003, 22, 869–883. [Google Scholar] [CrossRef][Green Version]
- Johnson, P. (National Cancer Institute, Frederick, MD, USA). Personal communication, 2008.
- Oh, W.J.; Rishi, V.; Orosz, A.; Gerdes, M.J.; Vinson, C. Inhibition of CCAAT/enhancer binding protein family DNA binding in mouse epidermis prevents and regresses papillomas. Cancer Res. 2007, 67, 1867–1876. [Google Scholar] [CrossRef]
- Wong, P.S.; Li, W.; Vogel, C.F.; Matsumura, F. Characterization of MCF mammary epithelial cells overexpressing the Arylhydrocarbon receptor (AhR). BMC Cancer 2009, 9, 234. [Google Scholar] [CrossRef]
- Vogel, C.F.; Matsumura, F. Interaction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with induced adipocyte differentiation in mouse embryonic fibroblasts (MEFs) involves tyrosine kinase c-Src. Biochem. Pharmacol. 2003, 7, 1231–1244. [Google Scholar] [CrossRef]
- Ng, S.; Yoshida, K.; Zelikoff, J.T. Tumor challenges in immunotoxicity testing. Methods Mol. Biol. 2010, 598, 143–155. [Google Scholar] [PubMed]
- Selig, W.; Tocker, J. Effect of interleukin-1 receptor antagonist on antigen-induced pulmonary responses in guinea pigs. Eur. J. Pharmacol. 1992, 213, 331–336. [Google Scholar] [CrossRef]
- Pyo, H.; Choy, H.; Amorino, G.P.; Kim, J.S.; Cao, Q.; Hercules, S.K.; DuBois, R.N. A selective cyclooxygenase-2 inhibitor, NS-398, enhances the effect of radiation in vitro and in vivo preferentially on the cells that express cyclooxygenase-2. Clin. Cancer Res. 2001, 10, 2998–3005. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | PKA Activity (pmol/min/mg Protein) | |||
|---|---|---|---|---|
| MEF wt | MEF AhRR Tg | |||
| basal | total | basal | total | |
| Control | 110 ± 20 | 2644 ± 150 | 95 ± 14 | 2450 ± 32 |
| TCDD | 290 ± 38 a | 3720 ± 240 a | 170 ± 25 b | 2865 ± 80 b |
| MNF+TCDD | 145 ± 28 c | 2830 ± 110 c | 126 ± 40 c | 2050 ± 90 c |
| CH223191+TCDD | 123 ± 21 c | 2540 ± 140 c | 102 ± 20 c | 1980 ± 110 c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogel, C.F.A.; Ishihara, Y.; Campbell, C.E.; Kado, S.Y.; Nguyen-Chi, A.; Sweeney, C.; Pollet, M.; Haarmann-Stemmann, T.; Tuscano, J.M. A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth. Cancers 2019, 11, 589. https://doi.org/10.3390/cancers11050589
Vogel CFA, Ishihara Y, Campbell CE, Kado SY, Nguyen-Chi A, Sweeney C, Pollet M, Haarmann-Stemmann T, Tuscano JM. A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth. Cancers. 2019; 11(5):589. https://doi.org/10.3390/cancers11050589
Chicago/Turabian StyleVogel, Christoph F. A., Yasuhiro Ishihara, Claire E. Campbell, Sarah Y. Kado, Aimy Nguyen-Chi, Colleen Sweeney, Marius Pollet, Thomas Haarmann-Stemmann, and Joseph M. Tuscano. 2019. "A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth" Cancers 11, no. 5: 589. https://doi.org/10.3390/cancers11050589
APA StyleVogel, C. F. A., Ishihara, Y., Campbell, C. E., Kado, S. Y., Nguyen-Chi, A., Sweeney, C., Pollet, M., Haarmann-Stemmann, T., & Tuscano, J. M. (2019). A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth. Cancers, 11(5), 589. https://doi.org/10.3390/cancers11050589