Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells

 and
and 
Abstract
1. Introduction
2. Results
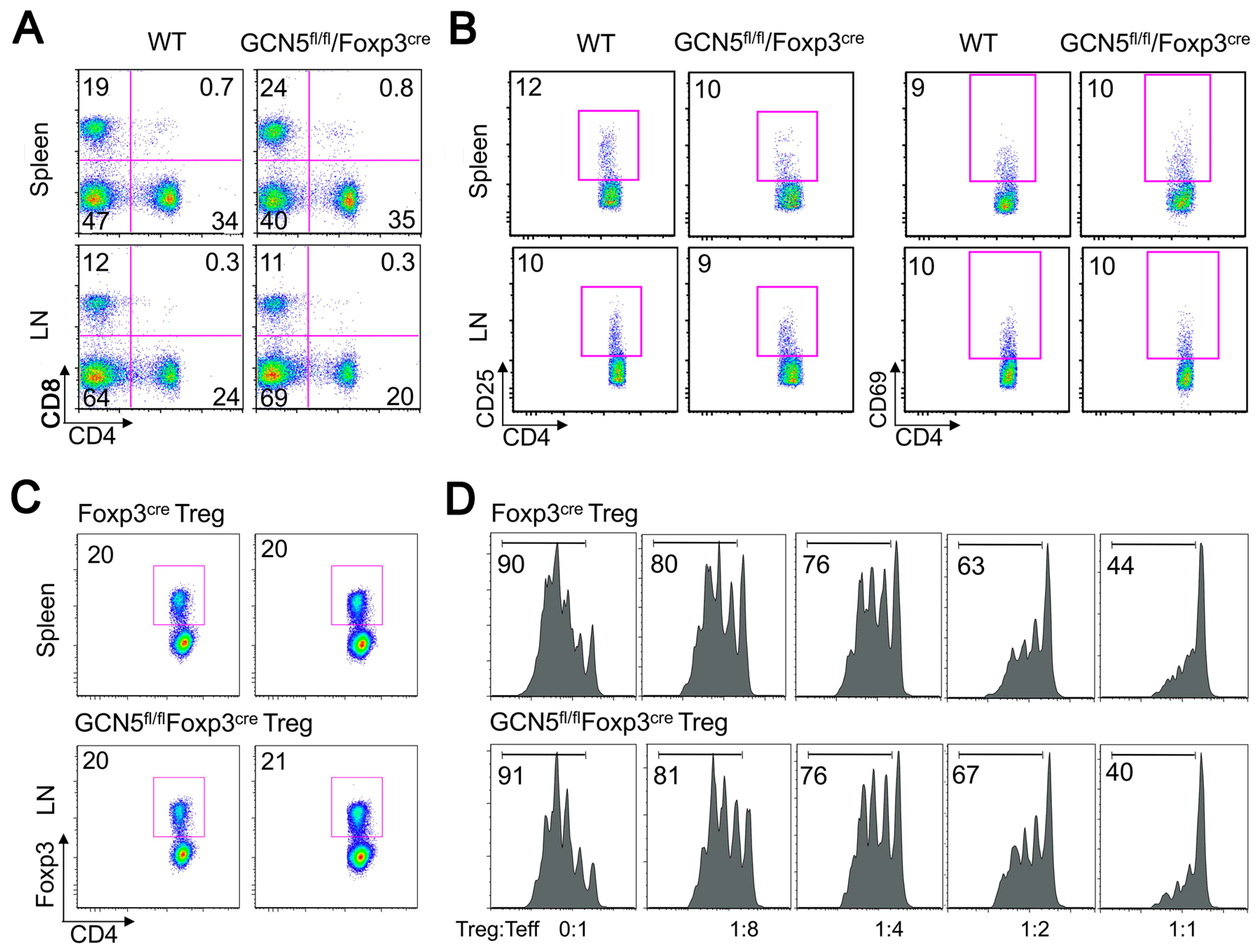
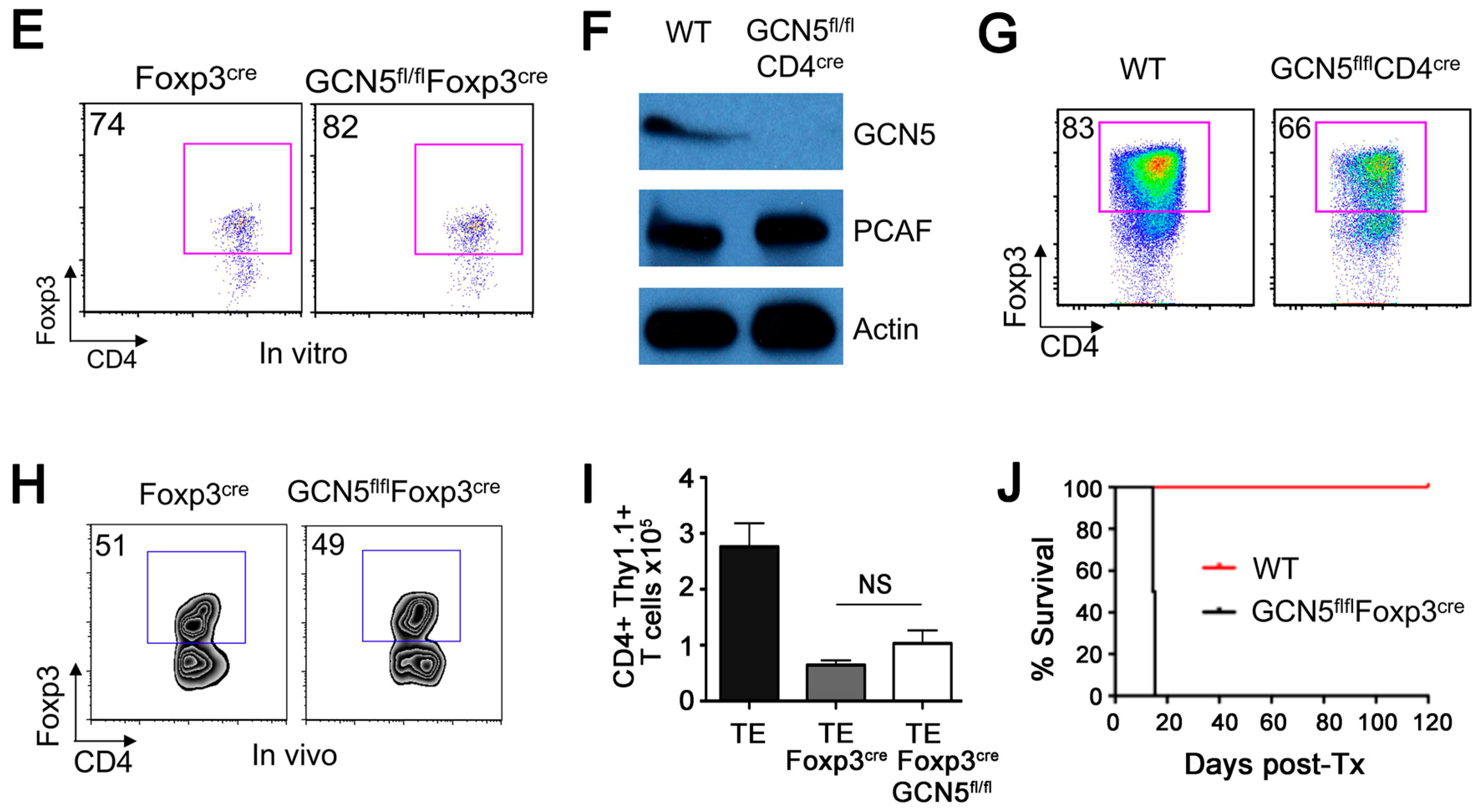
2.1. No Effects of GCN5 Deletion on Tregs In Vitro but Inhibitory Effects on Treg Function In Vivo
2.2. GCN5 Deletion Had No Effect on Overall T Cell Development but Impaired Teff Cell Functions
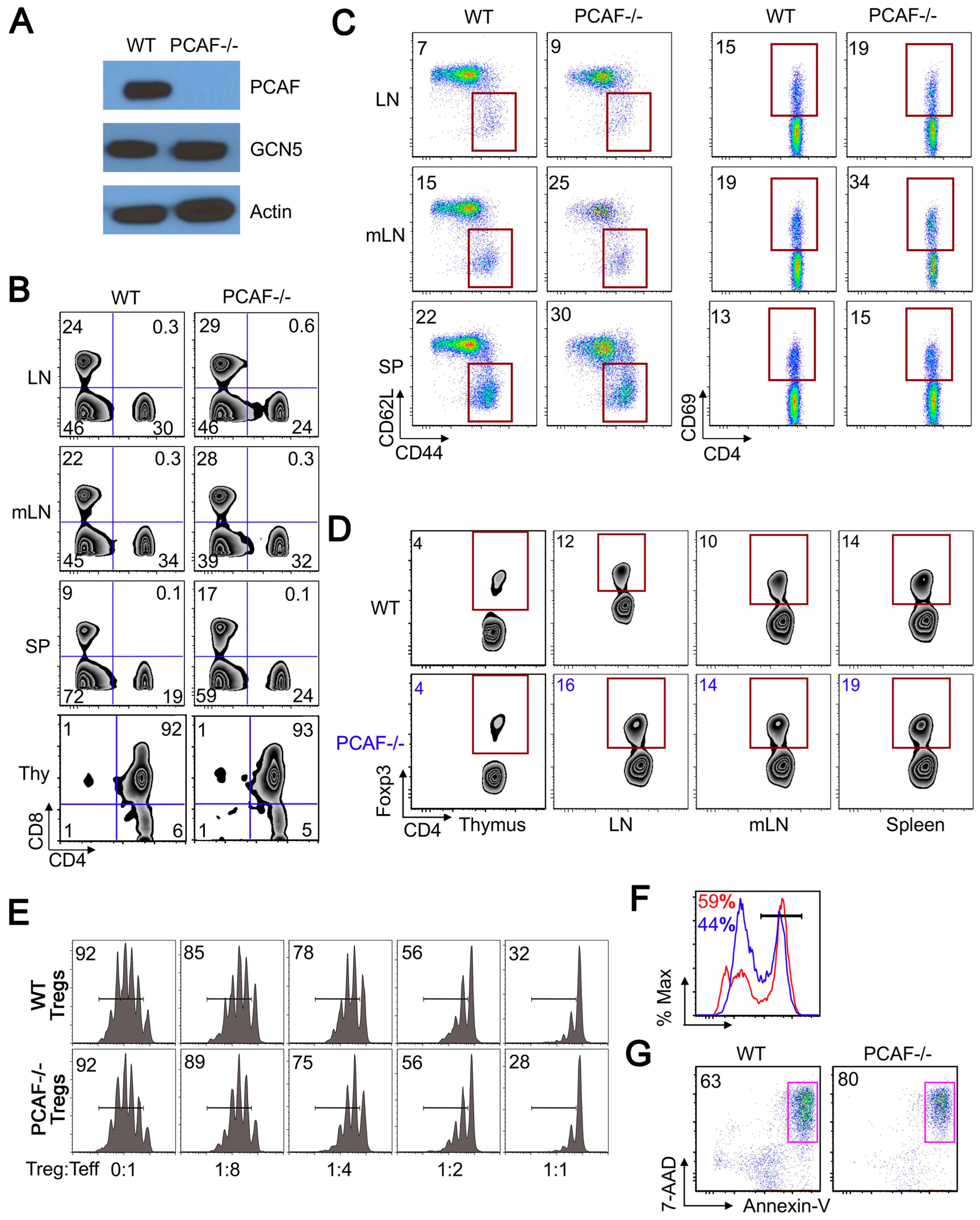
2.3. PCAF Helps Maintain Treg Stability Under TCR Stimulation
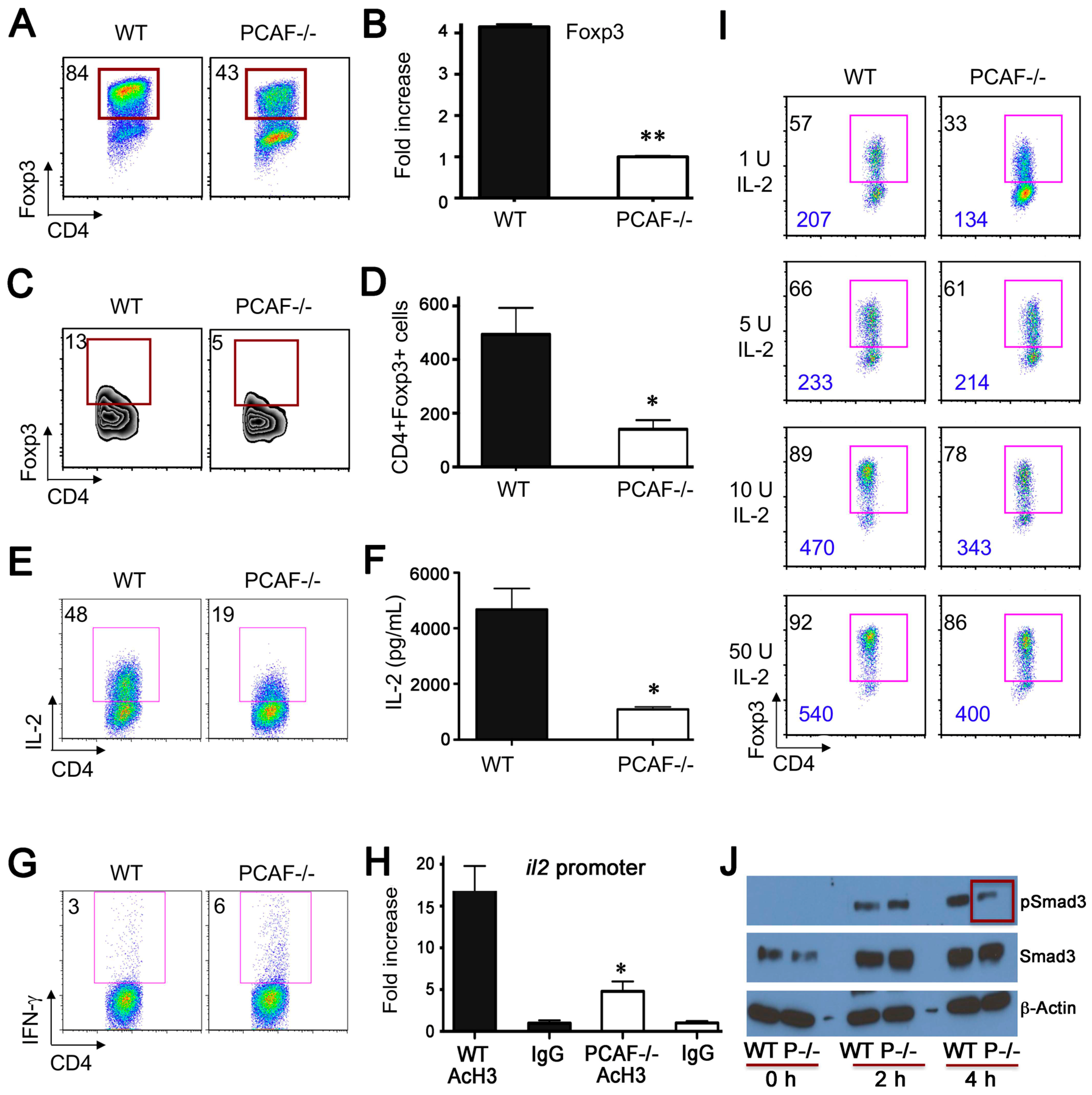
2.4. PCAF Is Important for iTreg Production
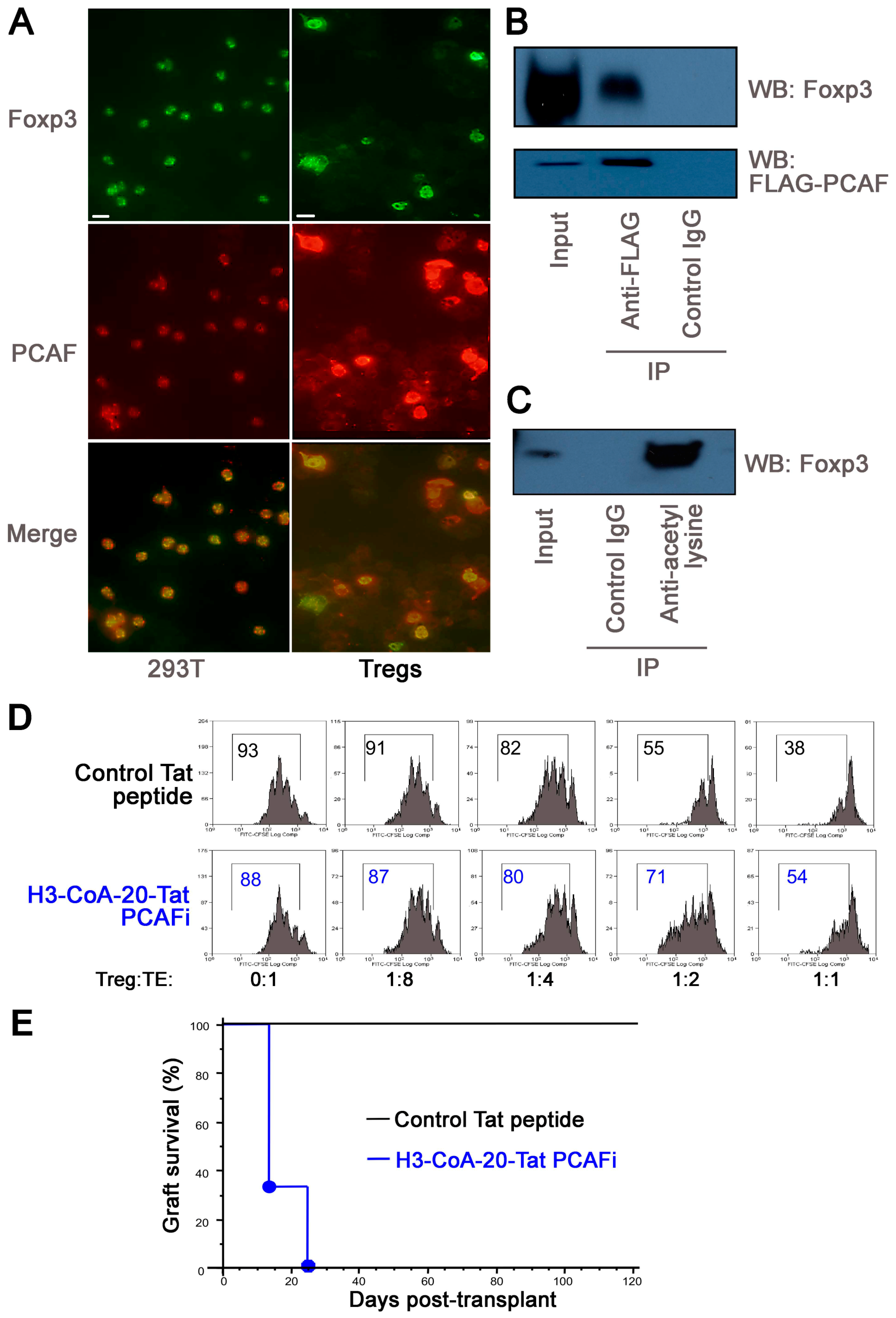
2.5. PCAF Targeting in WT Mice Impairs Inhibits Treg Function In Vitro and In Vivo
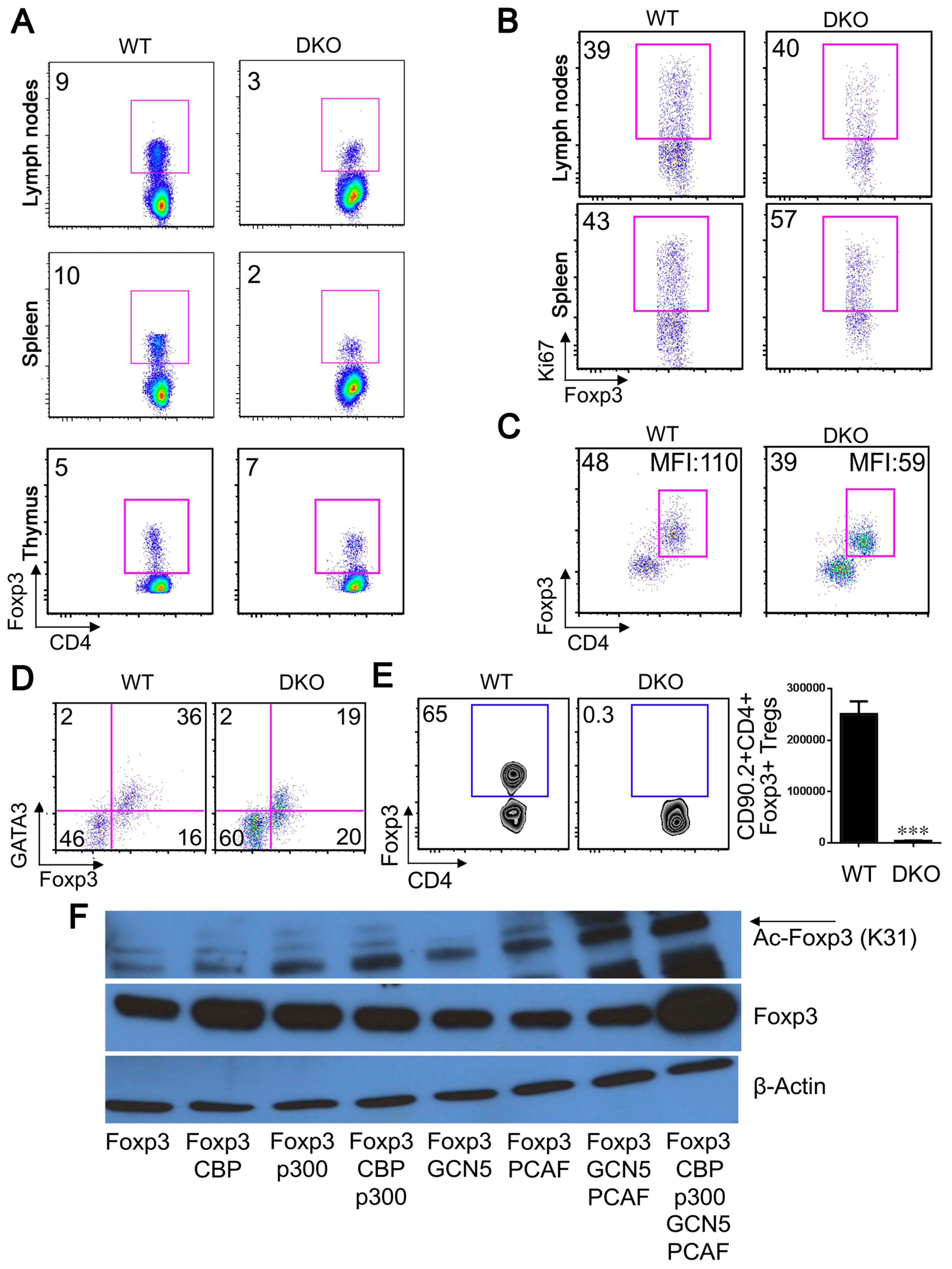
2.6. GCN5 and PCAF Deletion in nTregs Causes Lethal Autoimmunity
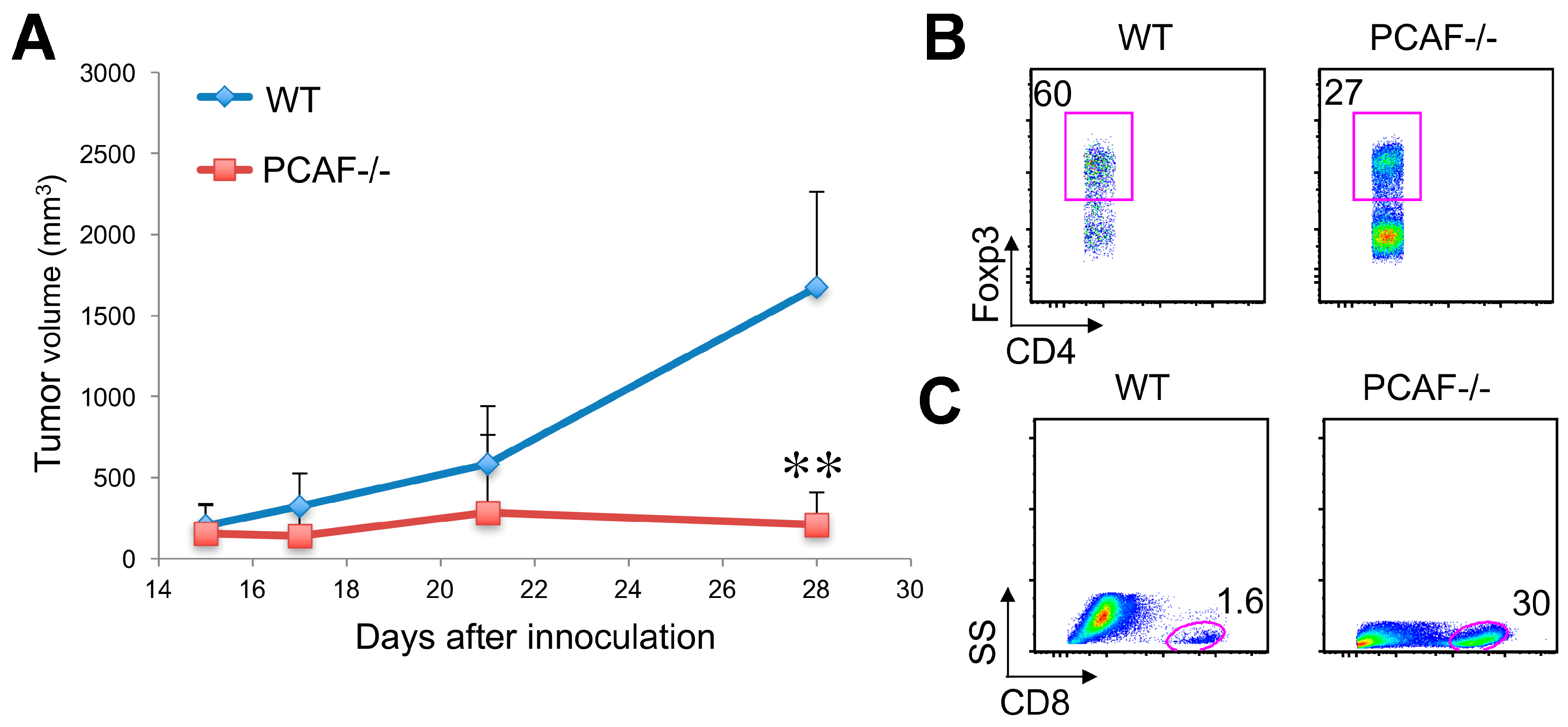
2.7. PCAF and Anti-Tumor Immunity
3. Discussion
4. Materials and Methods
4.1. Mice and Cardiac Allografting
4.2. Antibodies, Flow Cytometry and Cell Sorting
4.3. Plasmids
4.4. Treg Assays
4.5. Western Blotting
4.6. Quantitative PCR (qPCR) and ChIP Assays
4.7. Co-Localization Studies
4.8. Homeostatic Proliferation
4.9. Cardiac Transplant Studies
4.10. TC1 Tumor Growth
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DKO | double knockout |
| GCN5 | general control nonrepressed-protein 5 |
| GNAT | GCN5-related N-acetyltransferase |
| HAT | histone/protein acetyltransferase |
| iTreg | inducible Treg |
| KAT | lysine acetyltransferase |
| mAb | monoclonal antibody |
| nTreg | naturally occurring or thymic Treg |
| PCAF | p300/CBP-associated factor |
| Teff | T-effector cell |
| Treg | T-regulatory cell |
| WT | wild-type |
| YFP | Yellow fluorescent protein |
References
- Iizuka-Koga, M.; Nakatsukasa, H.; Ito, M.; Akanuma, T.; Lu, Q.; Yoshimura, A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J. Autoimmun. 2017, 83, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Min, B. Heterogeneity and stability in Foxp3+ regulatory T cells. J. Interferon Cytokine Res. 2017, 37, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef]
- Beier, U.H.; Wang, L.; Bhatti, T.R.; Liu, Y.; Han, R.; Ge, G.; Hancock, W.W. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol. Cell. Biol. 2011, 31, 1022–1029. [Google Scholar] [CrossRef]
- de Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.C.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef]
- Wang, L.; Kumar, S.; Dahiya, S.; Wang, F.; Wu, J.; Newick, K.; Han, R.; Samanta, A.; Beier, U.H.; Akimova, T.; et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. EBioMedicine 2016, 13, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, Y.; Cole, P.A.; Marmorstein, R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: Implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.; Wang, L.; Han, R.; Beier, U.H.; Akimova, T.; Bhatti, T.; Xiao, H.; Cole, P.A.; Brindle, P.K.; Hancock, W.W. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol. Cell. Biol. 2014, 34, 3993–4007. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Xiao, Y.; Nagai, Y.; Deng, G.; Ohtani, T.; Zhu, Z.; Zhou, Z.; Zhang, H.; Ji, M.Q.; Lough, J.W.; Samanta, A.; et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep. 2014, 7, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edmondson, D.G.; Roth, S.Y. Mammalian GCN5 and P/CAF acetyltransferases have homologous amino-terminal domains important for recognition of nucleosomal substrates. Mol. Cell. Biol. 1998, 18, 5659–5669. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edmondson, D.G.; Evrard, Y.A.; Wakamiya, M.; Behringer, R.R.; Roth, S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000, 26, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Yamauchi, J.; Kuwata, T.; Tamura, T.; Yamashita, T.; Bae, N.; Westphal, H.; Ozato, K.; Nakatani, Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 11303–11306. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Wang, C.; Kuang, X.; Feng, X.; Sartorelli, V.; Ying, H.; Ge, K.; Dent, S.Y. Gcn5 and PCAF regulate PPARgamma and Prdm16 expression to facilitate brown adipogenesis. Mol. Cell. Biol. 2014, 34, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Zhuang, L.; Lai, B.; Wang, C.; Li, W.; Dolan, B.; Lu, Y.; Wang, Z.; Zhao, K.; Peng, W.; et al. Gcn5 and PCAF negatively regulate interferon-beta production through HAT-independent inhibition of TBK1. EMBO Rep. 2014, 15, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Lohr, J.; Kahn, E.; Bluestone, J.A.; Abbas, A.K. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 2005, 202, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef]
- Itoh, S.; Ericsson, J.; Nishikawa, J.; Heldin, C.H.; ten Dijke, P. The transcriptional co-activator P/CAF potentiates TGF-beta/Smad signaling. Nucleic Acids Res. 2000, 28, 4291–4298. [Google Scholar] [CrossRef] [PubMed]
- Lau, O.D.; Kundu, T.K.; Soccio, R.E.; Ait-Si-Ali, S.; Khalil, E.M.; Vassilev, A.; Wolffe, A.P.; Nakatani, Y.; Roeder, R.G.; Cole, P.A. HATs off: Selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell 2000, 5, 589–595. [Google Scholar] [CrossRef]
- Lau, O.D.; Courtney, A.D.; Vassilev, A.; Marzilli, L.A.; Cotter, R.J.; Nakatani, Y.; Cole, P.A. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J. Biol. Chem. 2000, 275, 21953–21959. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.; Sitwala, K.V.; Khodadoust, M.S.; Kwok, R.P.; Mor-Vaknin, N.; Cebrat, M.; Cole, P.A.; Markovitz, D.M. p300/CBP-associated factor drives DEK into interchromatin granule clusters. J. Biol. Chem. 2005, 280, 31760–31767. [Google Scholar] [CrossRef]
- Wang, Y.; Su, M.A.; Wan, Y.Y. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity 2011, 35, 337–348. [Google Scholar] [CrossRef]
- Whiteside, T.L. FOXP3+ Treg as a therapeutic target for promoting anti-tumor immunity. Expert Opin. Ther. Targets 2018, 22, 353–363. [Google Scholar] [CrossRef]
- Akimova, T.; Hancock, W.W. How little is known about the role of human FOXP3+ Tregs in tumors. Expert Opin. Ther. Targets 2018, 22, 655–658. [Google Scholar] [CrossRef]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Wang, L.; Dent, S.Y.R. Functions of SAGA in development and disease. Epigenomics 2014, 6, 329–339. [Google Scholar] [CrossRef]
- Richters, A.; Koehler, A.N. Epigenetic modulation using small molecules—Targeting histone acetyltransferases in disease. Curr. Med. Chem. 2017, 24, 4121–4150. [Google Scholar] [CrossRef]
- Maurice, T.; Duclot, F.; Meunier, J.; Naert, G.; Givalois, L.; Meffre, J.; Celerier, A.; Jacquet, C.; Copois, V.; Mechti, N.; et al. Altered memory capacities and response to stress in p300/CBP-associated factor (PCAF) histone acetylase knockout mice. Neuropsychopharmacology 2008, 33, 1584–1602. [Google Scholar] [CrossRef] [PubMed]
- Bodai, L.; Pallos, J.; Thompson, L.M.; Marsh, J.L. Pcaf modulates polyglutamine pathology in a Drosophila model of Huntington’s disease. Neuro-Degenerative Dis. 2012, 9, 104–106. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, M.J.; Kim, Y.J.; Lee, Y.H.; Bae, D.; Kim, S.; Na, Y.; Yoon, H.G. Selective PCAF inhibitor ameliorates cognitive and behavioral deficits by suppressing NF-kappaB-mediated neuroinflammation induced by Abeta in a model of Alzheimer’s disease. Int. J. Mol. Med. 2015, 35, 1109–1118. [Google Scholar] [CrossRef]
- Song, X.; Li, B.; Xiao, Y.; Chen, C.; Wang, Q.; Liu, Y.; Berezov, A.; Xu, C.; Gao, Y.; Li, Z.; et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 2012, 1, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Beier, U.H.; Akimova, T.; Dahiya, S.; Han, R.; Samanta, A.; Levine, M.H.; Hancock, W.W. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am. J. Transplant. 2018, 18, 1596–1603. [Google Scholar] [CrossRef]
- Moustakim, M.; Clark, P.G.; Trulli, L.; Fuentes de Arriba, A.L.; Ehebauer, M.T.; Chaikuad, A.; Murphy, E.J.; Mendez-Johnson, J.; Daniels, D.; Hou, C.D.; et al. Discovery of a PCAF bromodomain chemical probe. Angew. Chem. 2017, 56, 827–831. [Google Scholar] [CrossRef]
- Lee, P.P.; Fitzpatrick, D.R.; Beard, C.; Jessup, H.K.; Lehar, S.; Makar, K.W.; Perez-Melgosa, M.; Sweetser, M.T.; Schlissel, M.S.; Nguyen, S.; et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001, 15, 763–774. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R., Jr.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, Z.; Srajer, G.; Chen, Y.C.; Huang, M.; Phan, H.M.; Dent, S.Y. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev. Dyn. 2008, 237, 928–940. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Han, R.; Beier, U.H.; Hancock, W.W. Two lysines in the forkhead domain of foxp3 are key to T regulatory cell function. PLoS ONE 2012, 7, e29035. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Han, R.; Beier, U.H.; Bhatti, T.R.; Akimova, T.; Greene, M.I.; Hiebert, S.W.; Hancock, W.W. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J. Clin. Investig. 2015, 125, 1111–1123. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Histology |
|---|---|
| Lungs | Lungs show moderate to marked perivascular and peribronchiolar lymphocytic infiltrates primarily centered around bronchovascular bundles and also extends to distal airspaces with widening of intra-alveolar septae. |
| Liver | Hepatic architecture is intact with mild to moderately dense lymphocytic infiltration involving the majority of large and small portal tracts. Inflammation is also present within the lobules and there is diffuse extramedullary hematopoiesis, characterized by scattered collections of erythroid precursors and occasional megakaryocytes, within sinusoids. |
| Spleen | Splenic parenchyma has accentuation and expansion of peri-arteriolar lymphoid sheaths. Collections of mature lymphocytes and foci of extramedullary hematopoiesis, characterized by numerous megakaryocytes and erythroid precursors, are present in the red pulp. |
| LN | Lymph nodes are cellular, but there is effacement of nodal architecture with loss of distinct follicles and germinal center formation is not seen. |
| Thymus | Thymic tissue is markedly diminished in volume and the corticomedullary junction is indistinct. Cortical thymocytes appear mature. |
| Skin | A diffuse mononuclear inflammatory infiltrate is predominantly composed of small mature lymphocytes within the superficial dermis, with focal exocytosis. |
| Brain | Brain features are normal without significant leptomeningeal or parenchymal inflammation. |
| Pancreas | Variable, mild lymphocytic inflammation is present focal around islets of Langerhans, and with infiltration into adjacent peri-pancreatic adipose tissues. |
| Heart | Normal cardiac myocytes and vasculature. |
| Kidney | Normal histology. |
| Gut | Small bowel mucosa has scattered collections of mononuclear cells in the lamina propria, without infiltration into surface epithelium, and villous architecture is intact |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Bao, C.; Wang, L.; Han, R.; Beier, U.H.; Akimova, T.; Cole, P.A.; Dent, S.Y.R.; Hancock, W.W. Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells. Cancers 2019, 11, 554. https://doi.org/10.3390/cancers11040554
Liu Y, Bao C, Wang L, Han R, Beier UH, Akimova T, Cole PA, Dent SYR, Hancock WW. Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells. Cancers. 2019; 11(4):554. https://doi.org/10.3390/cancers11040554
Chicago/Turabian StyleLiu, Yujie, Chunrong Bao, Liqing Wang, Rongxiang Han, Ulf H. Beier, Tatiana Akimova, Philip A. Cole, Sharon Y. R. Dent, and Wayne W. Hancock. 2019. "Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells" Cancers 11, no. 4: 554. https://doi.org/10.3390/cancers11040554
APA StyleLiu, Y., Bao, C., Wang, L., Han, R., Beier, U. H., Akimova, T., Cole, P. A., Dent, S. Y. R., & Hancock, W. W. (2019). Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells. Cancers, 11(4), 554. https://doi.org/10.3390/cancers11040554