The Transient Receptor Potential Vanilloid Type-2 (TRPV2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk between Transcription Factors and Signaling Molecules


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
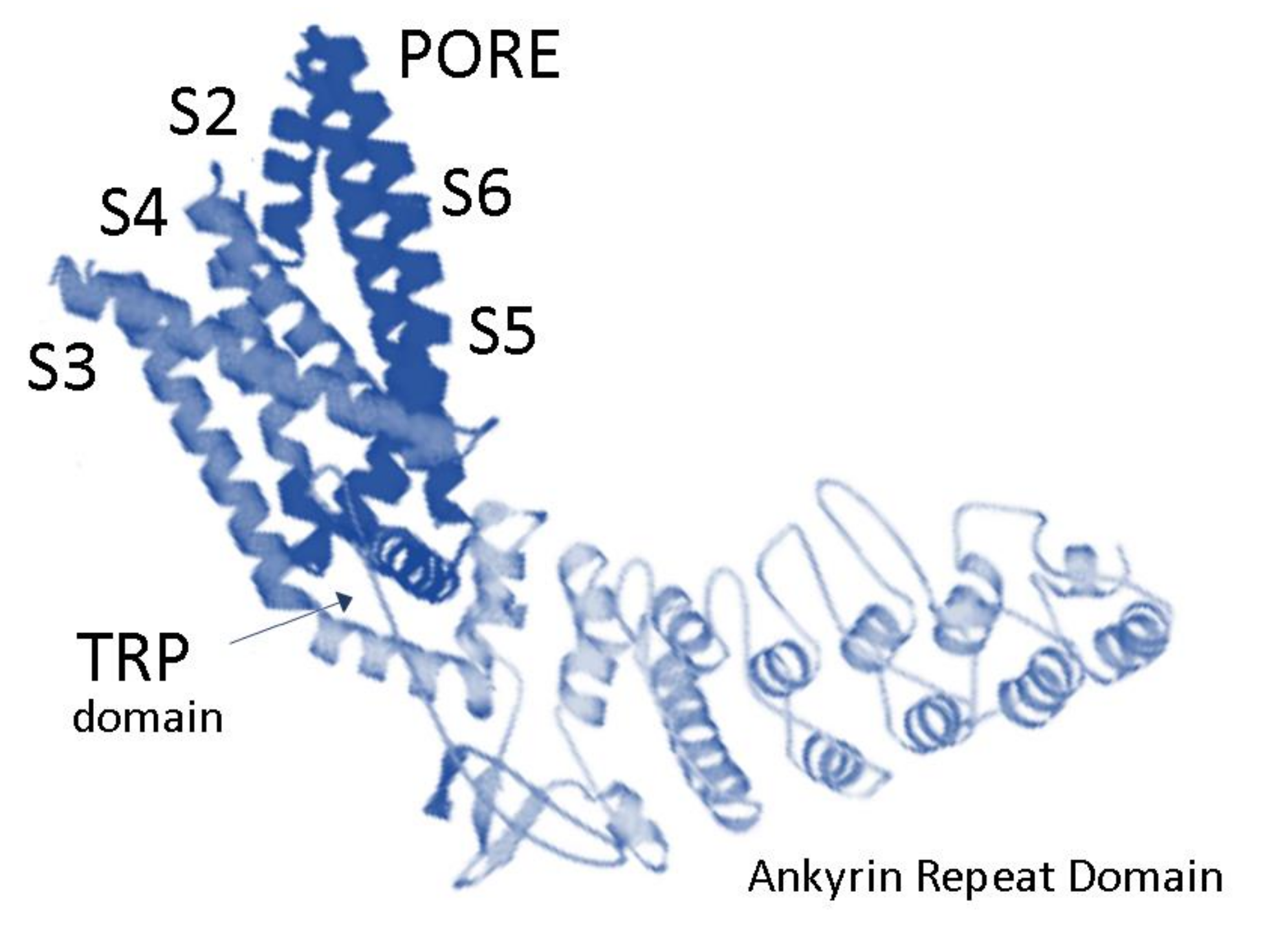
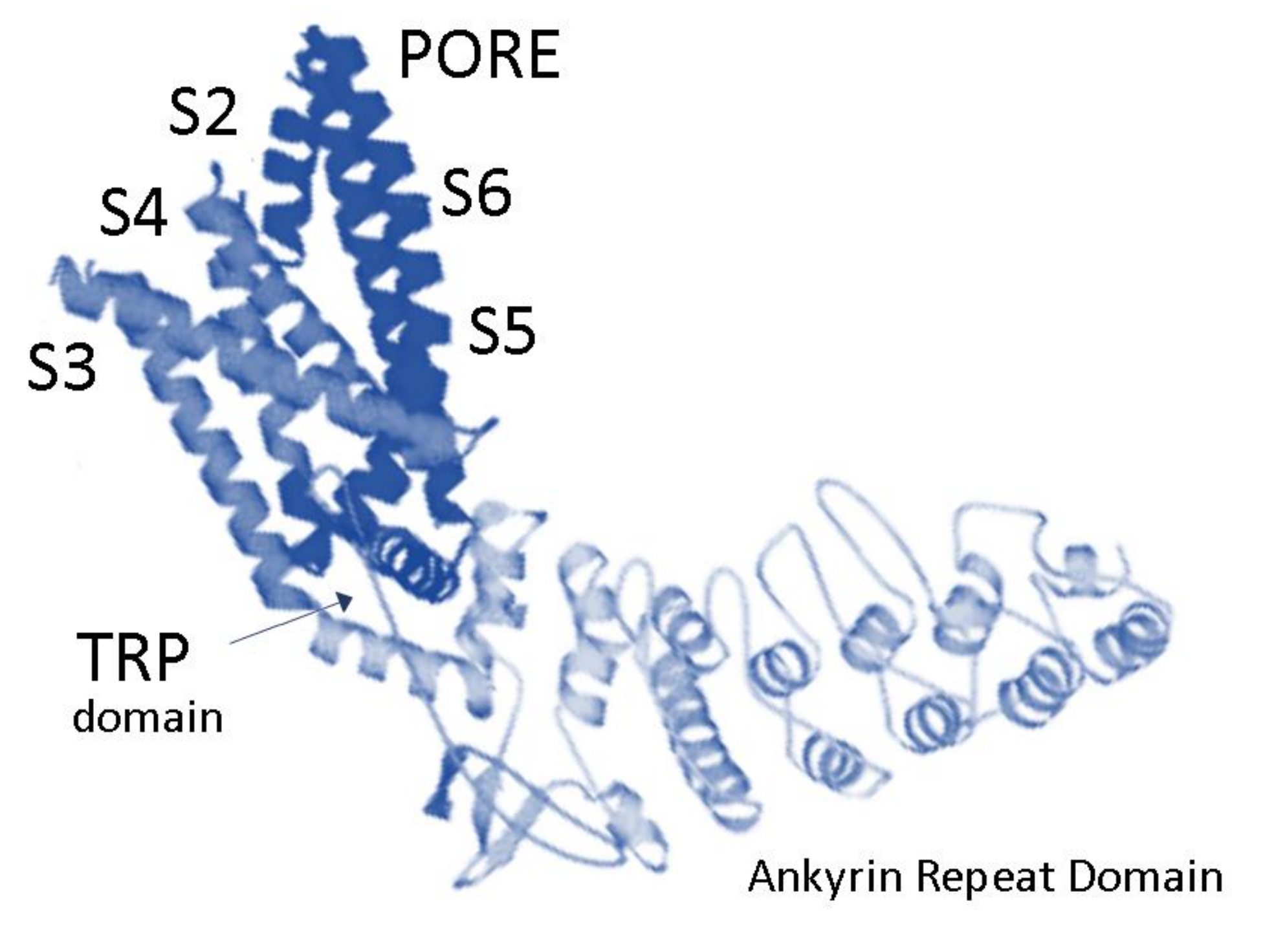
:1. Expression of TRPV2 in Mammalian Central and Peripheral Nervous Systems
2. Role of TRPV2 Ca2+ Channel in Developing Neurons and Its Regulation by Signaling Pathways
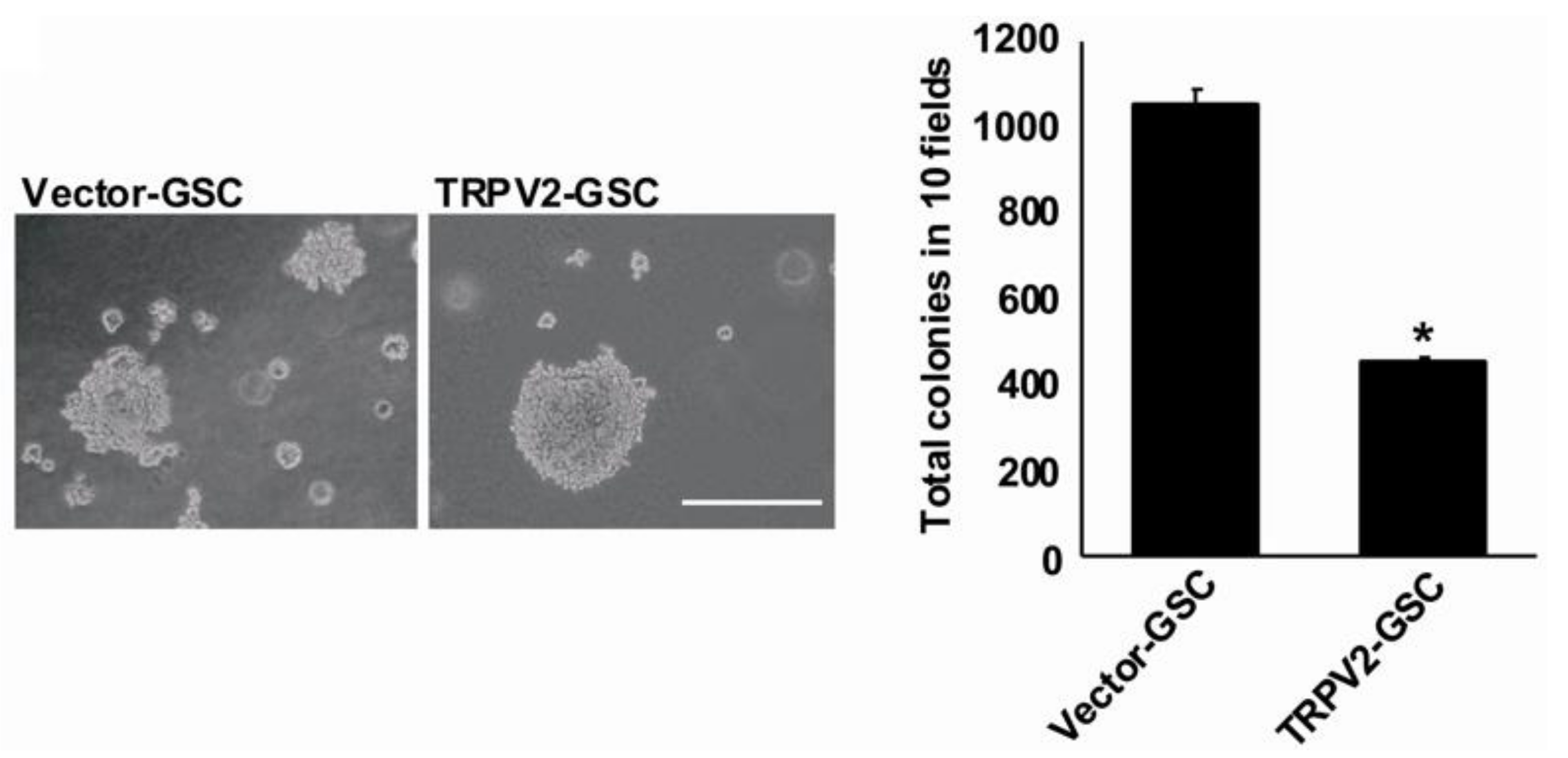
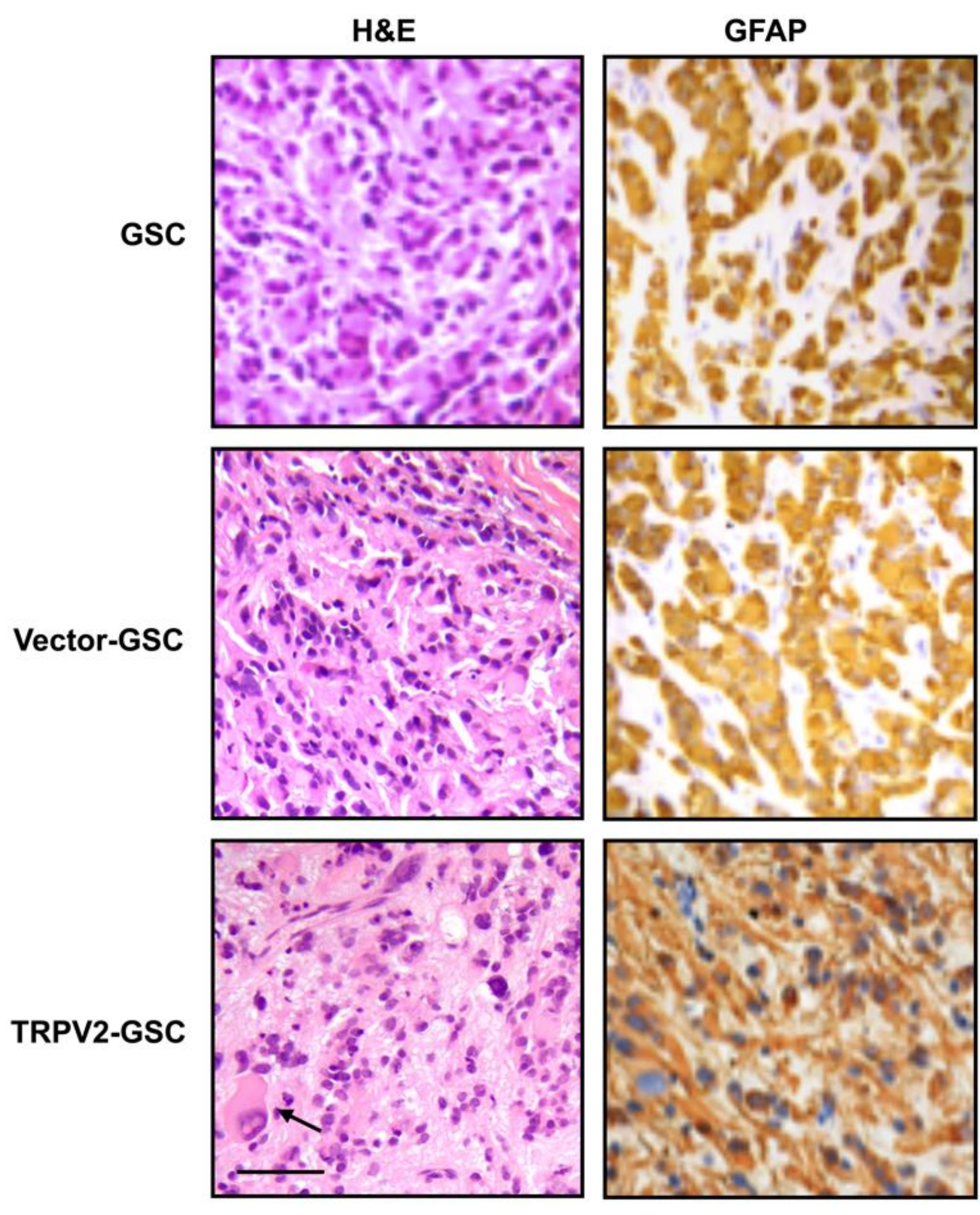
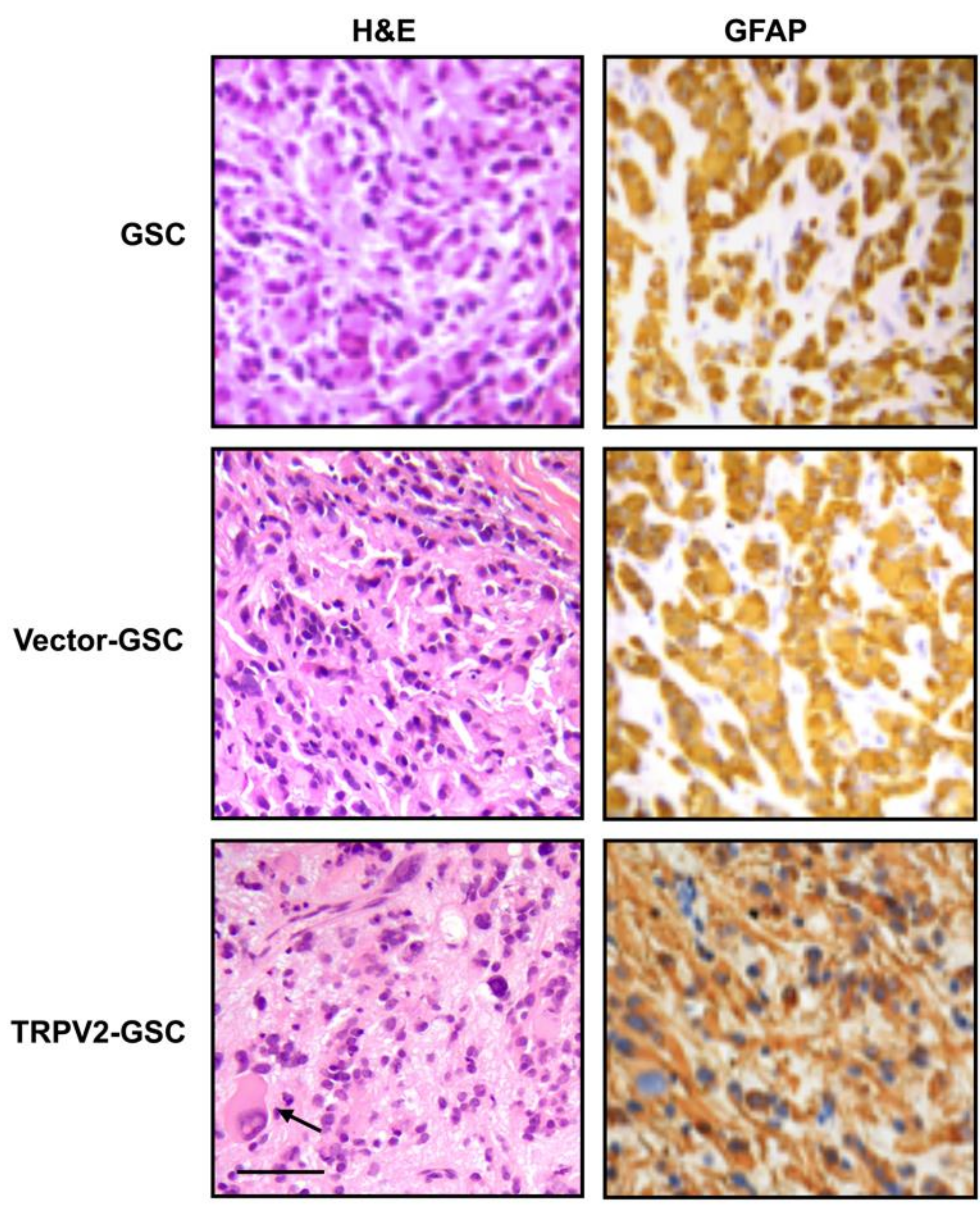
3. Expression and Function of TRPV2 Ca2+ Channels in Glioblastoma Stem/Progenitor-Like Cells (GSCs)
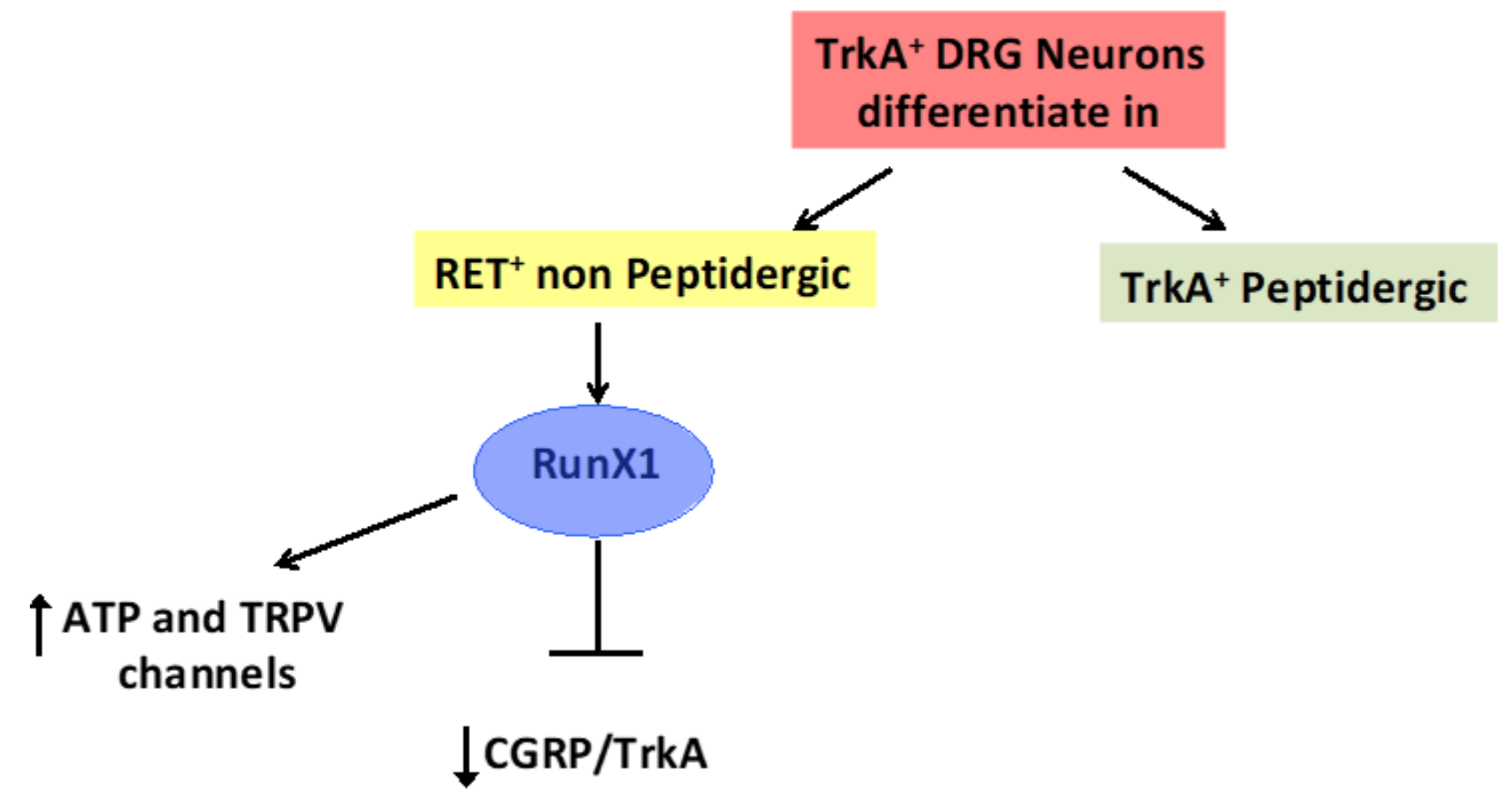
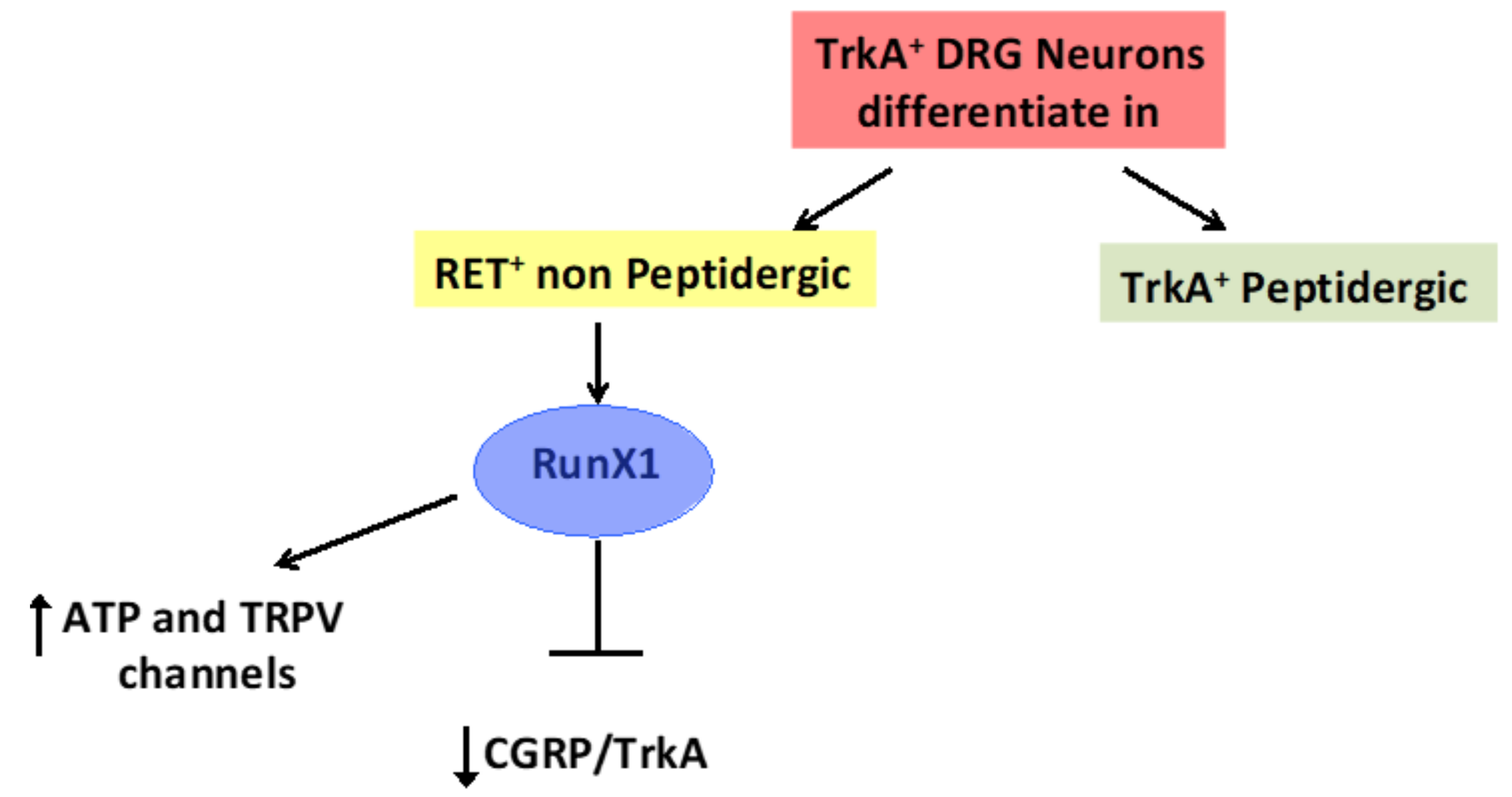
4. The Transcription Factor Aml1/Runx1 Regulates the Proliferation and Differentiation of GSCs
5. TRPV2 Channels in Glioblastoma Progression: TRPV2 Interactome-Based Signature as a Negative Prognostic Factor
6. Conclusion and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.W.; Cohen, M.R.; Jiang, J.; Samanta, A.; Lodowski, D.T.; Zhou, Z.H.; Moiseenkova-Bell, V.Y. Structure of the full-length TRPV2 channel by cryo-EM. Nat. Commun. 2016, 7, 11130. [Google Scholar] [CrossRef] [PubMed]
- Zubcevic, L.; Herzik, M.A., Jr.; Chung, B.C.; Liu, Z.; Lander, G.C.; Lee, S.Y. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Park, U.; Vastani, N.; Guan, Y.; Raja, S.N.; Koltzenburg, M.; Caterina, M.J. TRP vanilloid 2 knock-out mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J. Neurosci. 2011, 31, 11425–11436. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Hanson, P.I.; Schlesinger, P. Luminal chloride-dependent activation of endosome calcium channels: Patch clamp study of enlarged endosomes. J. Biol. Chem. 2007, 282, 27327–27333. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Puertollano, R. Role of TRP channels in the regulation of the endosomal pathway. Physiology (Bethesda) 2011, 26, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Ishizaki, Y.; Mandadi, S. Astrocytes express functional TRPV2 ion channels. Biochem. Biophys. Res. Commun. 2013, 441, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Nedungadi, T.P.; Dutta, M.; Bathina, C.S.; Caterina, M.J.; Cunningham, J.T. Expression and distribution of TRPV2 in rat brain. Exp. Neurol. 2012, 237, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Katanosaka, K.; Takatsu, S.; Mizumura, K.; Naruse, K.; Katanosaka, Y. TRPV2 is required for mechanical nociception and the stretch-evoked response of primary sensory neurons. Sci. Rep. 2018, 8, 6782. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Murayama, N.; Ono, K.; Ishizaki, Y.; Tominaga, M. TRPV2 enhances axon outgrowth through its activation by membrane stretch in developing sensory and motor neurons. J. Neurosci. 2010, 30, 4601–4612. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Taniguchi, K. Immunolocalization of VR1 and VRL1 in rat larynx. Auton. Neurosci. 2005, 117, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Lubin, M.L.; Flores, C.M. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J. Neurosci. 2008, 28, 6231–6238. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; di Martino, S.; Pallini, R.; Larocca, L.M.; Caprodossi, S.; Santoni, M.; et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int. J. Cancer 2012, 131, E1067–E1077. [Google Scholar] [CrossRef]
- Morgan, P.J.; Hübner, R.; Rolfs, A.; Frech, M.J. Spontaneous calcium transients in human neural progenitor cells mediated by transient receptor potential channels. Stem Cells Dev. 2013, 22, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.J.; Pujic, Z.; Goodhill, G.J. Calcium signaling in axon guidance. Trends Neurosci. 2014, 37, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.R.; Johnson, W.M.; Pilat, J.M.; Kiselar, J.; DeFrancesco-Lisowitz, A.; Zigmond, R.E.; Moiseenkova-Bell, V.Y. Nerve Growth Factor Regulates Transient Receptor Potential Vanilloid 2 via Extracellular Signal-Regulated Kinase Signaling to Enhance Neurite Outgrowth in Developing Neurons. Mol. Cell Biol. 2015, 35, 4238–4252. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Nagasawa, M.; Kojima, I.; Ishizaki, Y.; Shibasaki, K. Transient receptor potential vanilloid 2 activation by focal mechanical stimulation requires interaction with the actin cytoskeleton and enhances growth cone motility. FASEB J. 2017, 31, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xiao, J.; Hu, Z.; Xie, M.; Wang, W.; He, D. Blocking transient receptor potential vanilloid 2 channel in astrocytes enhances astrocyte-mediated neuroprotection after oxygen-glucose deprivation and reoxygenation. Eur. J. Neurosci. 2016, 44, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; Ginty, D.D. Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 2013, 14, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Penna, A.; Juvin, V.; Chemin, J.; Compan, V.; Monet, M.; Rassendren, F.A. PI3-kinase promotes TRPV2 activity independently of channel translocation to the plasma membrane. Cell Calcium 2006, 39, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, M.; Zhang, Y.Q.; Mashima, H.; Li, L.; Shibata, H.; Kojima, I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat. Cell Biol. 1999, 1, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Marlin, M.C.; Li, G. Biogenesis and function of the NGF/TrkA signaling endosome. Int. Rev. Cell Mol. Biol. 2015, 314, 239–257. [Google Scholar] [PubMed]
- Xue, Y.; Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Wang, Q.; Jin, C.; Zhou, Y.; Wen, L.; Ren, J. GPS 2.1: Enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011, 24, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Mobley, W.C. Signaling endosome hypothesis: A cellular mechanism for long distance communication. J. Neurobiol. 2004, 58, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C. Progenitor cells and glioma formation. Curr. Opin. Neurol. 2001, 14, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Dirks, P.B. Cancer: Stem cells and brain tumors. Nature 2006, 444, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Kang, S.K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [PubMed]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulam, R.; Alvarez, L.; Guzmán, M.; Galve-Roperh, I. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Q.; Yang, J.; Lou, M.; Wang, A.; Dong, J.; Qin, Z.; Zhang, T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res. 2010, 1313, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Schwartz, P.H.; Linskey, M.E.; Bota, D.A. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 2011, 76, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Binello, E.; Germano, I.M. Targeting glioma stem cells: A novel framework for brain tumors. Cancer Sci. 2011, 102, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Lytle, R.A.; Jiang, Z.; Zheng, X.; Rich, K.M. BCNU down- regulates anti-apoptotic proteins bcl-xL and Bcl-2 in association with cell death in oligodendroglioma-derived cells. J. Neurooncol. 2004, 68, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, S.J.; Chung, M.K.; Jung, K.H.; Choi, M.R.; Kim, Y.; Chai, Y.G.; Kim, S.J.; Park, K.S. High expression of large-conductance Ca2+-activated K+ channel in the CD133+ subpopulation of SH-SY5Y neuroblastoma cells. Biochem. Biophys. Res. Commun. 2010, 396, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.N.; Abrahamsen, B.; Baker, M.D.; Boorman, J.D.; Donier, E.; Drew, L.J.; Nassar, M.A.; Okuse, K.; Seereeram, A.; Stirling, C.L.; et al. Ion channel activities implicated in pathological pain. Novartis Found. Symp. 2004, 261, 32–40. [Google Scholar] [PubMed]
- Inoue, K.; Shiga, T.; Ito, Y. Runx transcription factors in neuronal development. Neural Dev. 2008, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Broom, D.C.; Liu, Y.; de Nooij, J.C.; Li, Z.; Cen, C.; Samad, O.A.; Jessell, T.M.; Woolf, C.J.; Ma, Q. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 2006, 49, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Theriault, F.M.; Nuthall, H.N.; Dong, Z.; Lo, R.; Barnabe-Heider, F.; Miller, F.D.; Stifani, S. Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J. Neurosci. 2005, 25, 2050–2061. [Google Scholar] [CrossRef] [PubMed]
- Theriault, F.M.; Roy, P.; Stifani, S. AML1/Runx1 is important for the development of hindbrain cholinergic branchiovisceral motor neurons and selected cranial sensory neurons. Proc. Natl. Acad. Sci. USA 2004, 101, 10343–10348. [Google Scholar] [CrossRef] [PubMed]
- Planagumà, J.; Díaz-Fuertes, M.; Gil-Moreno, A.; Abal, M.; Monge, M.; García, A.; Baró, T.; Thomson, T.M.; Xercavins, J.; Alameda, F.; et al. A differential gene expression profile reveals overexpression of RUNX1/AML1 in invasive endometrioid carcinoma. Cancer Res. 2004, 64, 8846–8853. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, C.; Hagiwara, A.; Miyagawa, K.; Nakashima, S.; Yoshikawa, T.; Kin, S.; Nakase, Y.; Ito, K.; Yamagishi, H.; Yazumi, S.; et al. Frequent downregulation of the runt domain transcription factors RUNX1, RUNX3 and their cofactor CBFB in gastric cancer. Int. J. Cancer 2005, 113, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; Sklan, E.H.; Birikh, K.; Shapira, M.; Trejo, L.; Eldor, A.; Soreq, H. Complex regulation of acetylcholinesterase gene expression in human brain tumors. Oncogene 2002, 21, 8428–8441. [Google Scholar] [CrossRef] [PubMed]
- Sangpairoj, K.; Vivithanaporn, P.; Apisawetakan, S.; Chongthammakun, S.; Sobhon, P.; Chaithirayanon, K. RUNX1 Regulates Migration, Invasion, and Angiogenesis via p38 MAPK Pathway in Human Glioblastoma. Cell Mol. Neurobiol. 2017, 37, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.J.; Yu, W.D.; Du, J.B.; Chao, S.; Chen, M.X.; Zhao, H.H.; Guo, J.Z. Overexpression or knock-down of runt-related transcription factor 1 affects BCR-ABL-induced proliferation and migration in vitro and leukemogenesis in vivo in mice. Chin. Med. J. (Engl.) 2009, 122, 331–337. [Google Scholar] [PubMed]
- Kurokawa, M.; Tanaka, T.; Tanaka, K.; Ogawa, S.; Mitani, K.; Yazaki, Y.; Hirai, H. Overexpression of the AML1 proto-oncoprotein in NIH3T3 cells leads to neoplastic transformation depending on the DNA-binding and transactivational potencies. Oncogene 1996, 12, 883–892. [Google Scholar] [PubMed]
- Michaud, J.; Simpson, K.M.; Escher, R.; Buchet-Poyau, K.; Beissbarth, T.; Carmichael, C.; Ritchie, M.E.; Schütz, F.; Cannon, P.; Liu, M.; et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genom. 2008, 9, 363. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Hong, D.; Gupta, R.; Matsuo, K.; Seto, M.; Enver, T. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007, 4, e172. [Google Scholar] [CrossRef] [PubMed]
- Coffman, J.A. Runx transcription factors and the developmental balance between cell proliferation and differentiation. Cell Biol. Int. 2003, 27, 315–324. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.S.; Ito, K.; Ito, Y. RUNX family: Regulation and diversification of roles through inter- acting proteins. Int. J. Cancer 2013, 132, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, Q. Generation of somatic sensory neuron diversity and implications on sensory coding. Curr. Opin. Neurobiol. 2011, 21, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kurokawa, M.; Yamaguchi, Y.; Izutsu, K.; Nitta, E.; Mitani, K.; Satake, M.; Noda, T.; Ito, Y.; Hirai, H. The corepressor mSin3A regulates phosphorylation-induced activation, intranuclear location, and stability of AML1. Mol. Cell Biol. 2004, 24, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Loilome, W.; Joshi, A.D.; Ap Rhys, C.M.; Piccirillo, S.; Vescovi, A.L.; Gallia, G.L.; Riggins, G.J. Glioblastoma cell growth is suppressed by disruption of Fibroblast Growth Factor pathway signaling. J. Neurooncol. 2009, 94, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Piirsoo, A.; Kasak, L.; Kauts, M.L.; Loog, M.; Tints, K.; Uusen, P.; Neuman, T.; Piirsoo, M. Protein kinase inhibitor SU6668 attenuates positive regulation of Gli proteins in cancer and multipotent progenitor cells. Biochim. Biophys. Acta 2014, 1843, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Wang, Q.; Lin, S.L.; Chang, C.D.; Chuang, J.; Ying, S.Y. Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp. Biol. Med. (Maywood) 2006, 231, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Song, Y.; Peng, Y.; Wang, H.; Long, H.; Yu, X.; Li, Z.; Fang, L.; Wu, A.; Luo, W.; et al. Zeb2 mediates multiple pathways regulating cell proliferation, migration, invasion, and apoptosis in glioma. PLoS ONE 2012, 7, e38842. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Puri, T.; Jha, P.; Pathak, P.; Joshi, N.; Suri, V.; Sharma, M.C.; Sharma, B.S.; Mahapatra, A.K.; Suri, A.; et al. A clinicopathological and molecular analysis of glioblastoma multiforme with long-term survival. J. Clin. Neurosci. 2011, 18, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R.; et al. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Chibon, F. Cancer gene expression signatures–The rise and fall? Eur. J. Cancer 2013, 49, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Ko, E.A.; Gu, W.; Lim, I.; Bang, H.; Zhou, T. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol. Cancer 2013, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Li, J.Y.; Liu, X.; Yan, X.Y.; Wang, W.; Wu, F.; Liang, T.Y.; Yang, F.; Hu, H.M.; Mao, H.X.; et al. A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget 2016, 7, 74895–74903. [Google Scholar] [CrossRef] [PubMed]
- Doñate-Macián, P.; Gómez, A.; Dégano, I.R.; Perálvarez-Marín, A. A TRPV2 interactome-based signature for prognosis in glioblastoma patients. Oncotarget 2018, 9, 18400–18409. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Mabuchi, T.; Satoh, E.; Furuya, K.; Zhang, L.; Maeda, S.; Naganuma, H. Expression of syndecans, a heparan sulfate proteoglycan, in malignant gliomas: Participation of nuclear factor-kappaB in upregulation of syndecan-1 expression. J. Neurooncol. 2006, 77, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Cooper, L.A.; Wang, F.; Gao, J.; Teodoro, G.; Scarpace, L.; Mikkelsen, T.; Schniederjan, M.J.; Moreno, C.S.; Saltz, J.H.; et al. Machine-based morphologic analysis of glioblastoma using whole-slide pathology images uncovers clinically relevant molecular correlates. PLoS ONE 2013, 8, e81049. [Google Scholar]
- Wang, L.; Wei, B.; Hu, G.; Wang, L.; Jin, Y.; Sun, Z. Gene expressionanalyses to explore the biomarkers and therapeutic targets for gliomas. Neurol. Sci. 2015, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.D.; Daneshvar, L.; Willert, J.R.; Matsumura, K.; Waldman, F.; Cogen, P.H. Physicalmapping of chromosome 17p13.3 in the region of a putative tumor suppressor gene important in medulloblastoma. Genomics 1994, 23, 229–322. [Google Scholar] [CrossRef] [PubMed]
- Willert, J.R.; Daneshvar, L.; Sheffield, V.C.; Cogen, P.H. Deletion of chromosomearm 17p DNA sequences in pediatric high-grade and juvenile pilocytic astrocytomas. Genes Chromosomes Cancer 1995, 12, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cheng, Q.; Ma, Z.; Xi, H.; Peng, R.; Jiang, B. Overexpression of RKIP inhibits cell invasion in glioma cell lines through upregulation of miR-98. Biomed. Res. Int. 2013, 2013, 695179. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.; Marie, S.K.; Oba-Shinjo, S.; Uno, M.; Izumi, C.; Oliveira, J.B.; Rosa, J.C. Quantitative protomi analysis shows differentially expressed HSPB1 in glioblastoma as a discriminating short from long serviva factor and NOVA1 as a differentiation factor between low-grade astrocytoma and oligodendroglioma. BMC Cancer 2015, 15, 481. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Chang, L.; Ye, W.; Jiang, C.; Zhang, Z. Raf kinase inhibitor protein (RKIP) inhibits the cell migration and invasion in human glioma cell linesin vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 14214–14220. [Google Scholar] [PubMed]
- Hsu, Y.C.; Kao, C.Y.; Chung, Y.F.; Lee, D.C.; Liu, J.W.; Chiu, I.M. Activation of Aurora A kinase through the FGF1/FGFR signaling axis sustains the stem cell characteristics of glioblastoma cells. Exp. Cell Res. 2016, 344, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Morrone, F.B.; Gehring, M.P.; Nicoletti, N.F. Calcium Channels and Associated Receptors in Malignant Brain Tumor Therapy. Mol. Pharmacol. 2016, 90, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Dall’Stella, P.B.; Docema, M.F.L.; Maldaun, M.V.C.; Feher, O.; Lancellotti, C.L.P. Case Report: Clinical Outcome and Image Response of Two Patients with Secondary High-Grade Glioma Treated with Chemoradiation, PCV, and Cannabidiol. Front. Oncol. 2019, 8, 643. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Katayama, Y.; Okuno, Y.; Wakabayashi, S. Novel inhibitor candidates of TRPV2 prevent damage of dystrophic myocytes and ameliorate against dilated cardiomyopathy in a hamster model. Oncotarget 2018, 9, 14042–14057. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoni, G.; Amantini, C. The Transient Receptor Potential Vanilloid Type-2 (TRPV2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk between Transcription Factors and Signaling Molecules. Cancers 2019, 11, 322. https://doi.org/10.3390/cancers11030322
Santoni G, Amantini C. The Transient Receptor Potential Vanilloid Type-2 (TRPV2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk between Transcription Factors and Signaling Molecules. Cancers. 2019; 11(3):322. https://doi.org/10.3390/cancers11030322
Chicago/Turabian StyleSantoni, Giorgio, and Consuelo Amantini. 2019. "The Transient Receptor Potential Vanilloid Type-2 (TRPV2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk between Transcription Factors and Signaling Molecules" Cancers 11, no. 3: 322. https://doi.org/10.3390/cancers11030322
APA StyleSantoni, G., & Amantini, C. (2019). The Transient Receptor Potential Vanilloid Type-2 (TRPV2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk between Transcription Factors and Signaling Molecules. Cancers, 11(3), 322. https://doi.org/10.3390/cancers11030322