The Novel Mnk1/2 Degrader and Apoptosis Inducer VNLG-152 Potently Inhibits TNBC Tumor Growth and Metastasis

 ,
, 
Abstract
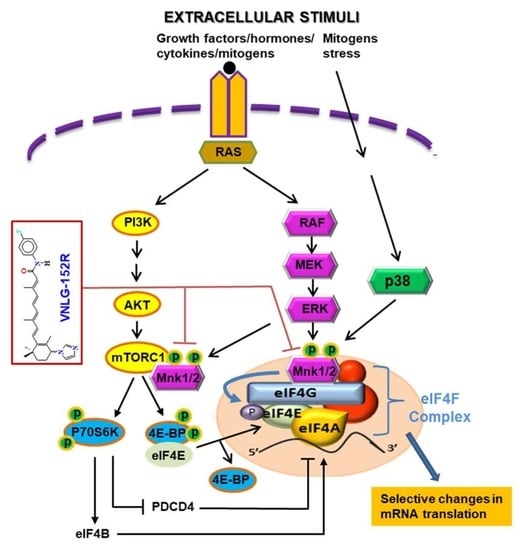
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. VNLG-152R Interacts with Mnk1, Inhibits eIF4E Complex Formation and − Is Important for Its Antiproliferative Activity in TNBC Cells in Vitro
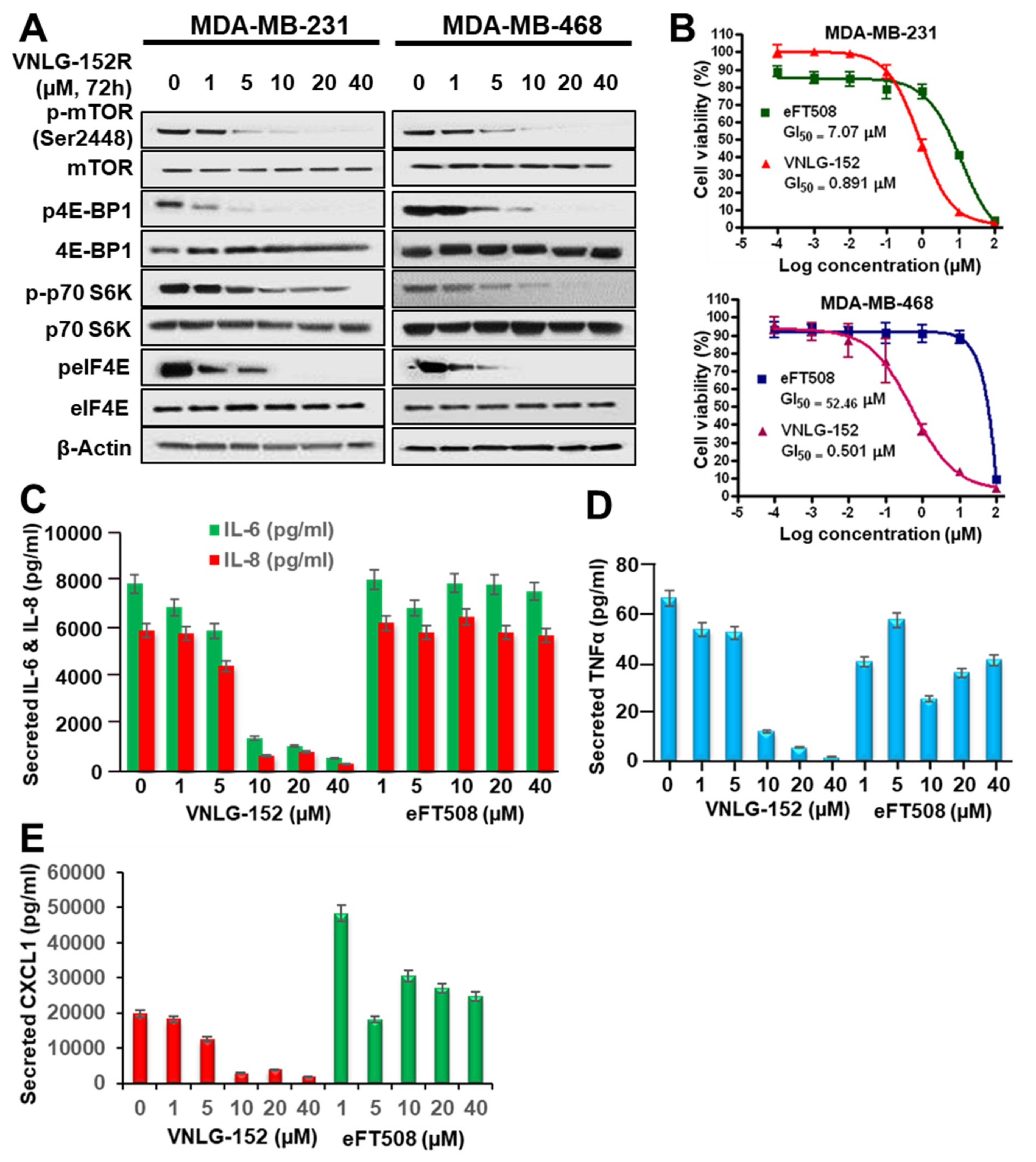
2.2. VNLG-152R Inhibits mTORC1 Signaling and the Production of Pro-Inflammatory Cytokines
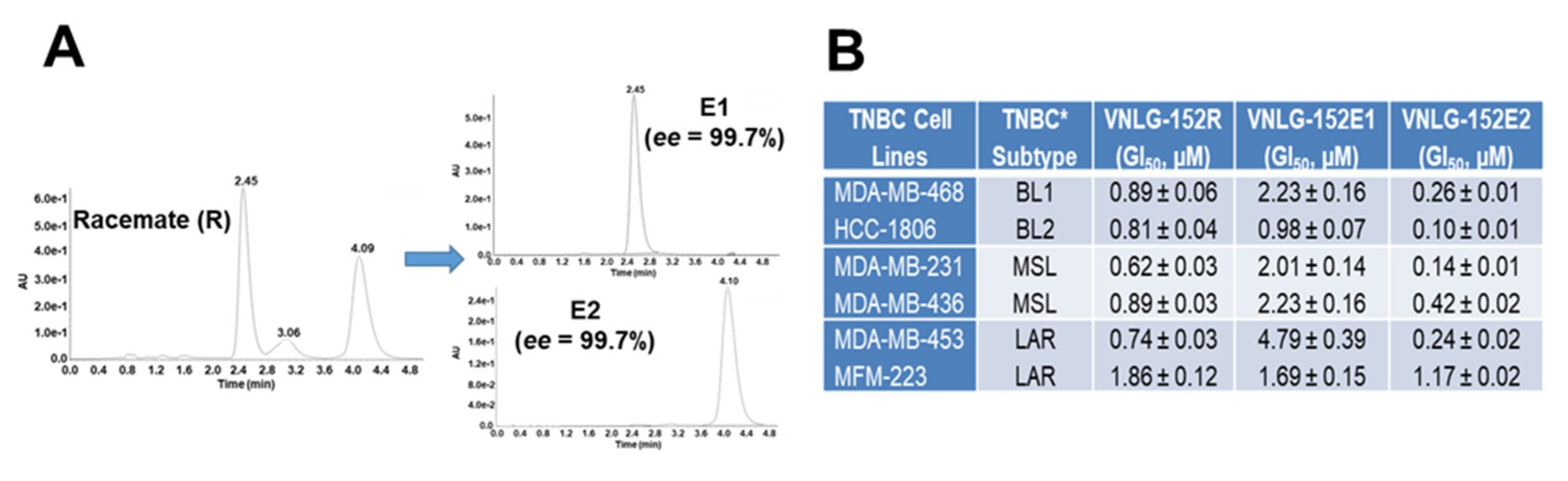
2.3. VNLG152R and Its Enantiomers Exhibit Differential Antiproliferative Activities and Pharmacokinetics but Exhibit Equivalent Safety/Toxicity Profiles
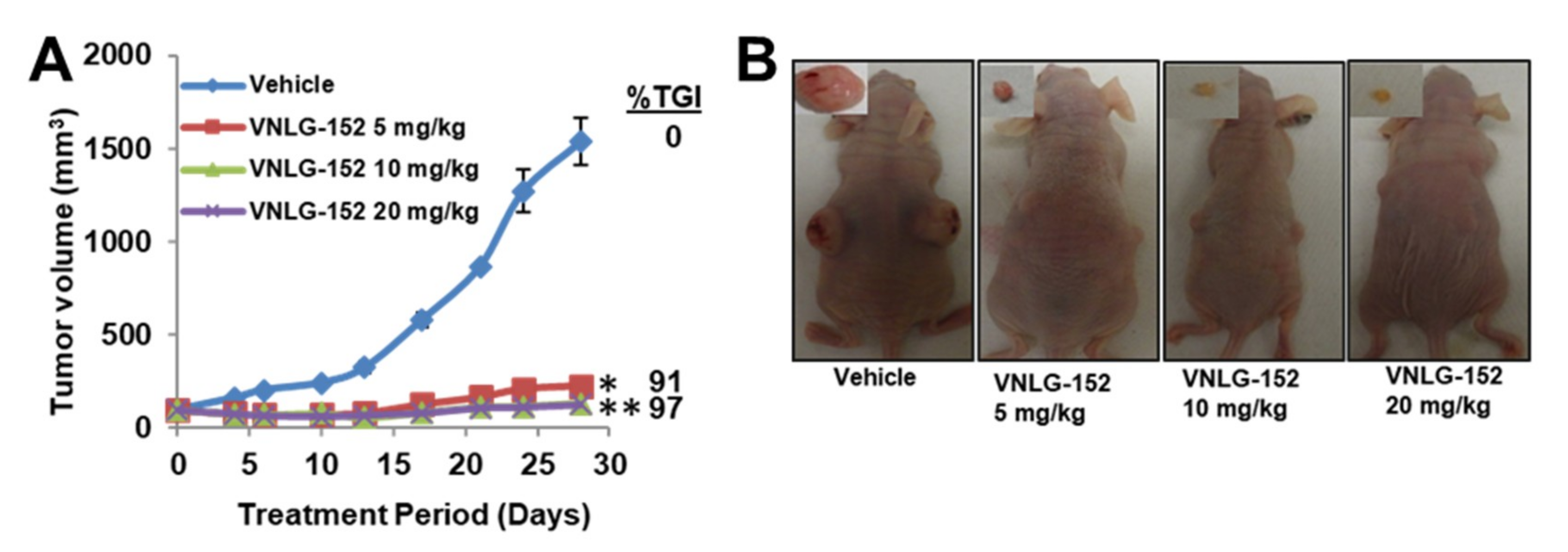
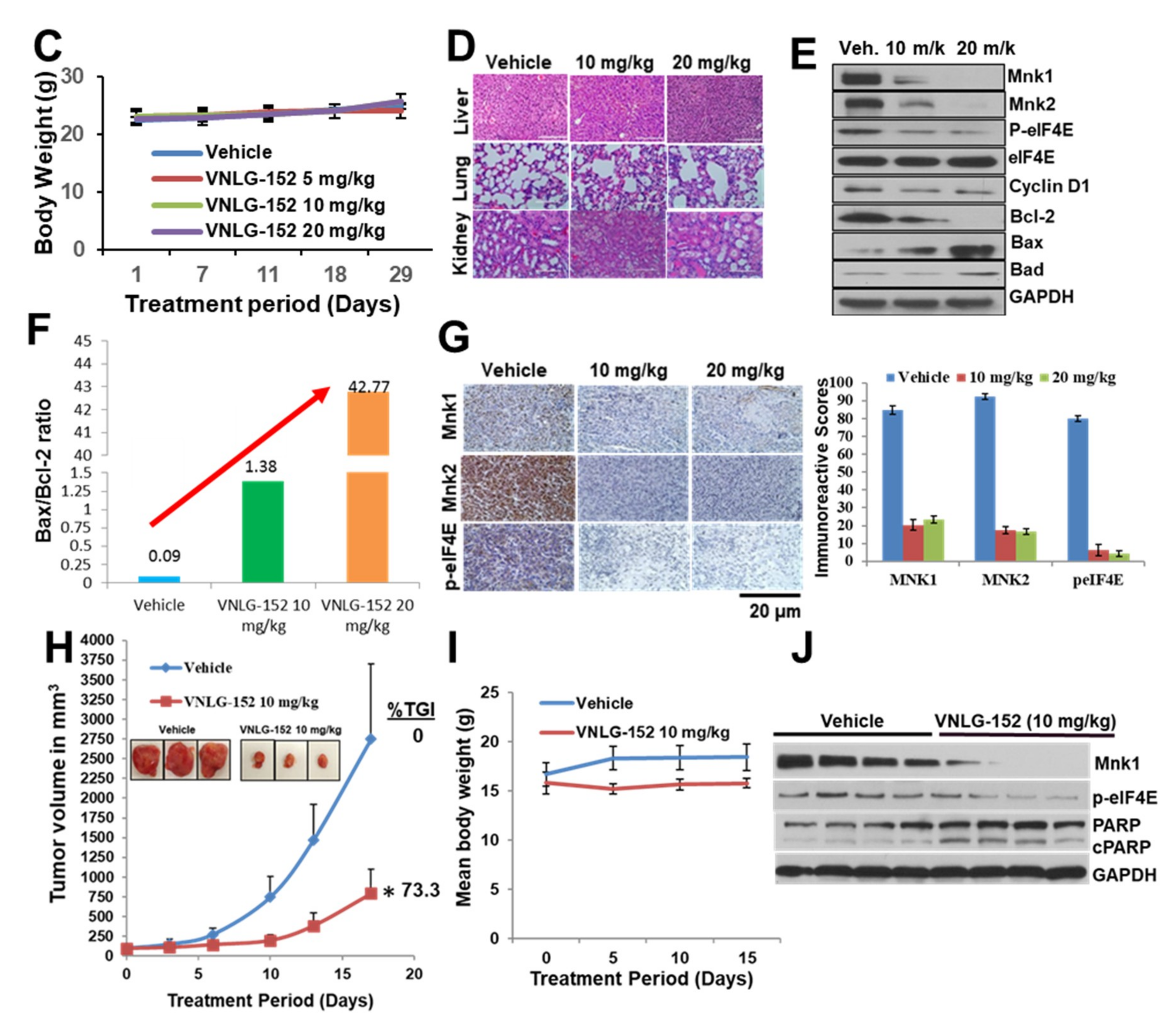
2.4. VNLG-152R Exhibited Potent Antitumor Activity in Cell-Line Derived and Patient Derived TNBC Xenograft Models and the Effect on Tumor Growth is Associated with Its Effects of Mnk1/2 and peIF4E, Downstream Targets and Induction of Apoptosis
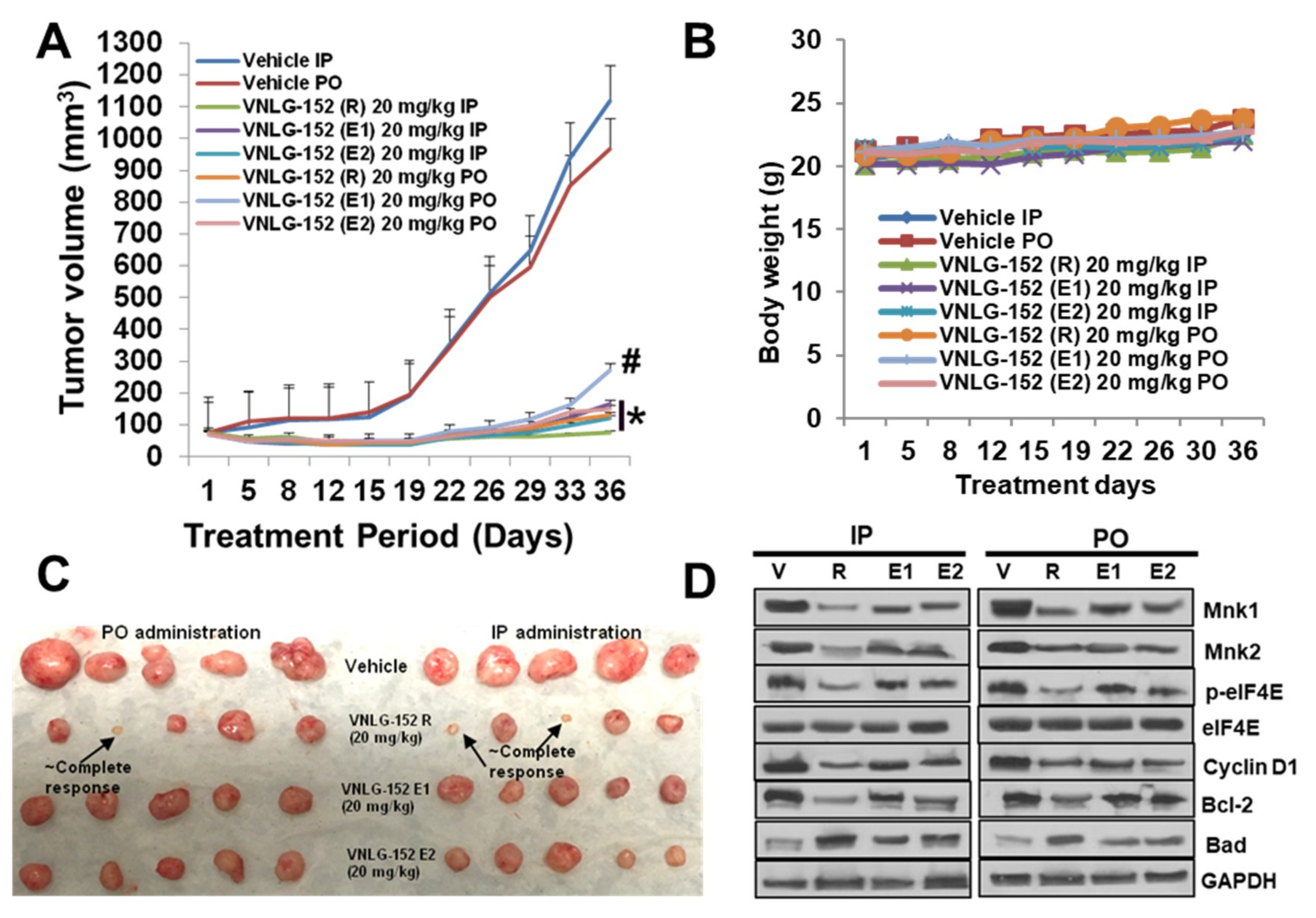
2.5. Racemic VNLG-152 Is Better than the Enantiomers at Inhibiting Tumor Growth and Mnk/eIF4E Signaling in TNBC Xenografts In Vivo
2.6. Racemic VNLG-152 Inhibits Cell Migration In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents.
4.2. Cell Culture Treatment and Western Blotting
4.3. Cell Growth Assays
4.4. In Vitro m7 GTP Pull-Down Assay
4.5. siRNA-Mediated Knockdown of Mnk1 Gene Expression
4.6. Real Time Cell Invasion Assay
4.7. Streptavidin–Agarose Pull-Down Assay
4.8. Multiplex Supernatant Cytokine Analysis (Luminex)
4.9. Surface Plasmon Resonance (SPR) Analysis
4.10. Animal Study Approval
4.10.1. In Vivo Anti-Tumor Studies in MDA-MB-231 TNBC Xenograft Model
4.10.2. In Vivo Anti-Tumor Studies in TNBC Patient Derived Xenograft (PDX) Tumor Model
4.10.3. Toxicology Study
4.10.4. In Vivo Experimental Lung Metastasis Assay
4.10.5. Immunohistochemical Analysis.
4.10.6. Tumor Lysate Preparation and Western Blot Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Vorobiof, D.A. Recent advances in the medical treatment of breast cancer. F1000Res 2016, 5, 2786. [Google Scholar] [CrossRef] [PubMed]
- Pareja, F.; Geyer, F.C.; Marchio, C.; Burke, K.A.; Weigelt, B.; Reis-Filho, J.S. Triple-negative breast cancer: The importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2016, 2, 16036. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Rida, P.; Ogden, A.; Ellis, I.O.; Varga, Z.; Wolff, A.C.; Traina, T.A.; Hatzis, C.; Palmer, J.R.; Ambrosone, C.B.; Lehmann, B.D.; et al. First international TNBC conference meeting report. Breast Cancer Res. Treat. 2018, 169, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.G.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, G.P.; Liu, C.; Zhou, M. Eukaryotic initiation factor 4E (eIF4E) and angiogenesis: Prognostic markers for breast cancer. BMC Cancer 2006, 6, 231. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N. eIF4E, the mRNA cap-binding protein: From basic discovery to translational research. Biochem. Cell Biol. 2008, 86, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Lachance, P.E.; Miron, M.; Raught, B.; Sonenberg, N.; Lasko, P. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol. Cell. Biol 2002, 22, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Akcakanat, A.; Singh, G.; Luyimbazi, D.; Zheng, Y.; Kim, D.; Gonzalez-Angulo, A.; Meric-Bernstam, F. eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling. Mol. Cancer Ther. 2008, 7, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, Y.; Qu, M.; Wan, H.; Cai, F.; Zhang, P. Inhibiting the MNK-eIF4E-beta-catenin axis increases the responsiveness of aggressive breast cancer cells to chemotherapy. Oncotarget 2017, 8, 2906–2915. [Google Scholar] [CrossRef] [PubMed]
- Wheater, M.J.; Johnson, P.W.; Blaydes, J.P. The role of MNK proteins and eIF4E phosphorylation in breast cancer cell proliferation and survival. Cancer Biol. Ther. 2010, 10, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Flowers, A.; Chu, Q.D.; Panu, L.; Meschonat, C.; Caldito, G.; Lowery-Nordberg, M.; Li, B.D. Eukaryotic initiation factor 4E overexpression in triple-negative breast cancer predicts a worse outcome. Surgery 2009, 146, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.O.; Carter, P.; Liu, L.; Li, B.D.; Abreo, F.; Tudor, A.; Zimmer, S.G.; De Benedetti, A. Elevated expression of eIF4E and FGF-2 isoforms during vascularization of breast carcinomas. Oncogene 1997, 15, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, N.; Del Rincon, S.V.; Huor, B.; Alain, T.; Petruccelli, L.A.; Hearnden, J.; Goncalves, C.; Grotegut, S.; Spruck, C.H.; Furic, L.; et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 2014. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.; White, S.; Chu, Q.; Meschonat, C.; Yu, H.; Johnson, L.W.; Debenedetti, A.; Abreo, F.; Turnage, R.H.; McDonald, J.C.; et al. High eIF4E, VEGF, and microvessel density in stage I to III breast cancer. Ann. Surg. 2006, 243, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Li, B.D.; Liu, L.; Dawson, M.; De Benedetti, A. Overexpression of eukaryotic initiation factor 4E (eIF4E) in breast carcinoma. Cancer 1997, 79, 2385–2390. [Google Scholar] [CrossRef]
- Li, B.D.; McDonald, J.C.; Nassar, R.; De Benedetti, A. Clinical outcome in stage I to III breast carcinoma and eIF4E overexpression. Ann. Surg. 1998, 227, 756–76l; discussion 761–763. [Google Scholar] [CrossRef] [PubMed]
- Lineham, E.; Tizzard, G.J.; Coles, S.J.; Spencer, J.; Morley, S.J. Synergistic effects of inhibiting the MNK-eIF4E and PI3K/AKT/ mTOR pathways on cell migration in MDA-MB-231 cells. Oncotarget 2018, 9, 14148–14159. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Diez, C.; Garcia-Recio, E.M.; Perez-Morgado, M.I.; Garcia-Hernandez, M.; Sanz-Criado, L.; Sacristan, S.; Toledo-Lobo, M.V.; Perez-Mies, B.; Esteban-Rodriguez, I.; Pascual, A.; et al. Increased expression of MNK1b, the spliced isoform of MNK1, predicts poor prognosis and is associated with triple-negative breast cancer. Oncotarget 2018, 9, 13501–13516. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.H.; Sprengeler, P.A.; Chiang, G.G.; Appleman, J.R.; Chen, J.; Clarine, J.; Eam, B.; Ernst, J.T.; Han, Q.; Goel, V.K.; et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J. Med. Chem. 2018, 61, 3516–3540. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, F.; Del Rincon, S.V.; Emond, A.; Huor, B.; Ngan, E.; Ng, J.; Dobocan, M.C.; Siegel, P.M.; Miller, W.H., Jr. Genetic and pharmacologic inhibition of eIF4E reduces breast cancer cell migration, invasion, and metastasis. Cancer Res. 2015, 75, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, F.; Del Rincon, S.V.; Miller, W.H., Jr. Eukaryotic translation initiation factor 4E as a novel therapeutic target in hematological malignancies and beyond. Expert Opin. Ther. Targets 2014, 18, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, F.; Yau, C.; Dobocan, M.C.; Culjkovic-Kraljacic, B.; Retrouvey, H.; Puckett, R.; Flores, L.M.; Krop, I.E.; Rousseau, C.; Cocolakis, E.; et al. Ribavirin treatment effects on breast cancers overexpressing eIF4E, a biomarker with prognostic specificity for luminal B-type breast cancer. Clin. Cancer Res. 2011, 17, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.P.; Ramalingam, S.; Kwegyir-Afful, A.K.; Hussain, A.; Njar, V.C. Targeting of protein translation as a new treatment paradigm for prostate cancer. Curr. Opin. Oncol. 2017, 29, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Dreas, A.; Mikulski, M.; Milik, M.; Fabritius, C.H.; Brzozka, K.; Rzymski, T. Mitogen-activated Protein Kinase (MAPK) Interacting Kinases 1 and 2 (MNK1 and MNK2) as Targets for Cancer Therapy: Recent Progress in the Development of MNK Inhibitors. Curr. Med. Chem. 2017, 24, 3025–3053. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Gromeier, M. MNK Controls mTORC1:Substrate Association through Regulation of TELO2 Binding with mTORC1. Cell Rep. 2017, 18, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Gromeier, M. MNK inversely regulates TELO2 vs. DEPTOR to control mTORC1 signaling. Mol. Cell. Oncol. 2017, 4, e1306010. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.K.; Njar, V.C.O. The retinamide VNLG-152 inhibits f-AR/AR-V7 and MNK-eIF4E signaling pathways to suppress EMT and castration-resistant prostate cancer xenograft growth. FEBS J. 2018, 285, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Beauchamp, E.; Bell, J.; Iqbal, A.; Su, B.; Fukunaga, R.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Regulatory effects of a Mnk2-eIF4E feedback loop during mTORC1 targeting of human medulloblastoma cells. Oncotarget 2014, 5, 8442–8451. [Google Scholar] [CrossRef] [PubMed]
- Grzmil, M.; Huber, R.M.; Hess, D.; Frank, S.; Hynx, D.; Moncayo, G.; Klein, D.; Merlo, A.; Hemmings, B.A. MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J. Clin. Investig. 2014, 124, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef] [PubMed]
- Teo, T.; Yu, M.; Yang, Y.; Gillam, T.; Lam, F.; Sykes, M.J.; Wang, S. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer Lett. 2015, 357, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Mbatia, H.W.; Ramalingam, S.; Ramamurthy, V.P.; Martin, M.S.; Kwegyir-Afful, A.K.; Njar, V.C. Novel C-4 heteroaryl 13-cis-retinamide Mnk/AR degrading agents inhibit cell proliferation and migration and induce apoptosis in human breast and prostate cancer cells and suppress growth of MDA-MB-231 human breast and CWR22Rv1 human prostate tumor xenografts in mice. J. Med. Chem. 2015, 58, 1900–1914. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Gediya, L.; Kwegyir-Afful, A.K.; Ramamurthy, V.P.; Purushottamachar, P.; Mbatia, H.; Njar, V.C. First MNKs degrading agents block phosphorylation of eIF4E, induce apoptosis, inhibit cell growth, migration and invasion in triple negative and Her2-overexpressing breast cancer cell lines. Oncotarget 2014, 5, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.; Kwegyir-Afful, A.K.; Njar, V.C. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget 2015, 6, 3195–3210. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Sasaki, M.; Elia, A.J.; Chio, II; Hamada, K.; Fukunaga, R.; Mak, T.W. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc. Natl. Acad. Sci. USA 2010, 107, 13984–13990. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Mnks, eIF4E phosphorylation and cancer. Biochim. Biophys. Acta 2015, 1849, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Dobashi, Y.; Watanabe, Y.; Miwa, C.; Suzuki, S.; Koyama, S. Mammalian target of rapamycin: A central node of complex signaling cascades. Int. J. Clin. Exp. Pathol. 2011, 4, 476–495. [Google Scholar] [PubMed]
- Santag, S.; Siegel, F.; Wengner, A.M.; Lange, C.; Bomer, U.; Eis, K.; Puhler, F.; Lienau, P.; Bergemann, L.; Michels, M.; et al. BAY 1143269, a novel MNK1 inhibitor, targets oncogenic protein expression and shows potent anti-tumor activity. Cancer Lett. 2017, 390, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- FDA’s policy statement for the development of new stereoisomeric drugs. Chirality 1992, 4, 338–340. [CrossRef] [PubMed]
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 1, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Agranat, I.; Wainschtein, S.R. The strategy of enantiomer patents of drugs. Drug Discov. Today 2010, 15, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Caner, H.; Groner, E.; Levy, L.; Agranat, I. Trends in the development of chiral drugs. Drug Discov. Today 2004, 9, 105–110. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Smith, S.W. Chiral toxicology: it’s the same thing...only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24 (Suppl. 2), S36–S40. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book ASCO Am. Soc. Clin. Oncol. Meet. 2015, e31–e39. [Google Scholar] [CrossRef] [PubMed]
- Bollag, W. Therapy of epithelial tumors with an aromatic retinoic acid analog. Chemotherapy 1975, 21, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Li, Y.; Yue, P.; Khuri, F.R.; Sun, S.Y. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia 2010, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Sun, M.; Yan, D.; Chen, K. Clinical significance of mTOR and eIF4E expression in invasive ductal carcinoma. Tumori 2014, 100, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Kim, R.H.; Sun, G.; Miller, J.K.; Li, B.D. Overexpression of eukaryotic initiation factor 4E is correlated with increased risk for systemic dissemination in node-positive breast cancer patients. J. Am. Coll. Surg. 2014, 218, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Bryant, J.D.; Dobrikova, E.Y.; Shveygert, M.; Bradrick, S.S.; Chandramohan, V.; Bigner, D.D.; Gromeier, M. Induction of viral, 7-methyl-guanosine cap-independent translation and oncolysis by mitogen-activated protein kinase-interacting kinase-mediated effects on the serine/arginine-rich protein kinase. J. Virol. 2014, 88, 13135–13148. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Platanias, L.C. Mnk Kinases in Cytokine Signaling and Regulation of Cytokine Responses. Biomol. Concepts 2012, 3, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhou, B.; Yang, C.Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Platanias, L.C. Mnk kinase pathway: Cellular functions and biological outcomes. World J. Biol. Chem. 2014, 5, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Diab, S.; Kumarasiri, M.; Yu, M.; Teo, T.; Proud, C.; Milne, R.; Wang, S. MAP kinase-interacting kinases—Emerging targets against cancer. Chem. Biol. 2014, 21, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Guo, J.; Yang, W.; Goncalves, C.; Rzymski, T.; Dreas, A.; Zylkiewicz, E.; Mikulski, M.; Brzozka, K.; Golas, A.; et al. MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J. Clin. Investig. 2017, 127, 4179–4192. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Cajal, S.R.Y.; Sonenberg, N.; Pelletier, J. Eukaryotic initiation factor 4F-sidestepping resistance mechanisms arising from expression heterogeneity. Curr. Opin. Genet. Dev. 2018, 48, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Ramon, Y.C.S.; Castellvi, J.; Hummer, S.; Peg, V.; Pelletier, J.; Sonenberg, N. Beyond molecular tumor heterogeneity: Protein synthesis takes control. Oncogene 2018, 37, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Ramon, Y.C.S.; De Mattos-Arruda, L.; Sonenberg, N.; Cortes, J.; Peg, V. The intra-tumor heterogeneity of cell signaling factors in breast cancer: p4E-BP1 and peIF4E are diffusely expressed and are real potential targets. Clin. Transl. Oncol. 2014, 16, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Beggs, J.E.; Tian, S.; Jones, G.G.; Xie, J.; Iadevaia, V.; Jenei, V.; Thomas, G.; Proud, C.G. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem. J. 2015, 467, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, N.; Hsu, B.E.; Istomine, R.; Alvarez, F.; Blagih, J.; Ma, E.H.; Morales, S.V.; Dai, D.L.; Li, G.; Souleimanova, M.; et al. Translational control in the tumor microenvironment promotes lung metastasis: Phosphorylation of eIF4E in neutrophils. Proc. Natl. Acad. Sci. USA 2018, 115, E2202–E2209. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Kosciuczuk, E.M.; Saleiro, D.; Platanias, L.C. Dual targeting of eIF4E by blocking MNK and mTOR pathways in leukemia. Cytokine 2017, 89, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Lineham, E.; Spencer, J.; Morley, S.J. Dual abrogation of MNK and mTOR: A novel therapeutic approach for the treatment of aggressive cancers. Future Med. Chem. 2017, 9, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Gau, Y.; Sabnis, G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res. Treat. 2014, 143, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K. Analysis of protein-DNA binding by streptavidin-agarose pulldown. Methods Mol. Biol. 2006, 338, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Lindamood, C., 3rd; Giles, H.D.; Hill, D.L. Preliminary toxicity profile of arotinoids SMR-2 and SMR-6 in male B6D2F1 mice. Fundam. Appl. Toxicol. 1987, 8, 517–530. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Voruganti, S.; Srivenugopal, K.S.; Nag, S.; Patil, S.; Sharma, H.; Wang, M.H.; Wang, H.; Buolamwini, J.K.; et al. The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models. Nat. Commun. 2014, 5, 5086. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramalingam, S.; Ramamurthy, V.P.; Gediya, L.K.; Murigi, F.N.; Purushottamachar, P.; Huang, W.; Choi, E.Y.; Zhang, Y.; Vasaitis, T.S.; Kane, M.A.; et al. The Novel Mnk1/2 Degrader and Apoptosis Inducer VNLG-152 Potently Inhibits TNBC Tumor Growth and Metastasis. Cancers 2019, 11, 299. https://doi.org/10.3390/cancers11030299
Ramalingam S, Ramamurthy VP, Gediya LK, Murigi FN, Purushottamachar P, Huang W, Choi EY, Zhang Y, Vasaitis TS, Kane MA, et al. The Novel Mnk1/2 Degrader and Apoptosis Inducer VNLG-152 Potently Inhibits TNBC Tumor Growth and Metastasis. Cancers. 2019; 11(3):299. https://doi.org/10.3390/cancers11030299
Chicago/Turabian StyleRamalingam, Senthilmurugan, Vidya P. Ramamurthy, Lalji K. Gediya, Francis N. Murigi, Puranik Purushottamachar, Weiliang Huang, Eun Yong Choi, Yuji Zhang, Tadas S Vasaitis, Maureen A. Kane, and et al. 2019. "The Novel Mnk1/2 Degrader and Apoptosis Inducer VNLG-152 Potently Inhibits TNBC Tumor Growth and Metastasis" Cancers 11, no. 3: 299. https://doi.org/10.3390/cancers11030299
APA StyleRamalingam, S., Ramamurthy, V. P., Gediya, L. K., Murigi, F. N., Purushottamachar, P., Huang, W., Choi, E. Y., Zhang, Y., Vasaitis, T. S., Kane, M. A., Lapidus, R. G., & Njar, V. C. O. (2019). The Novel Mnk1/2 Degrader and Apoptosis Inducer VNLG-152 Potently Inhibits TNBC Tumor Growth and Metastasis. Cancers, 11(3), 299. https://doi.org/10.3390/cancers11030299