Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
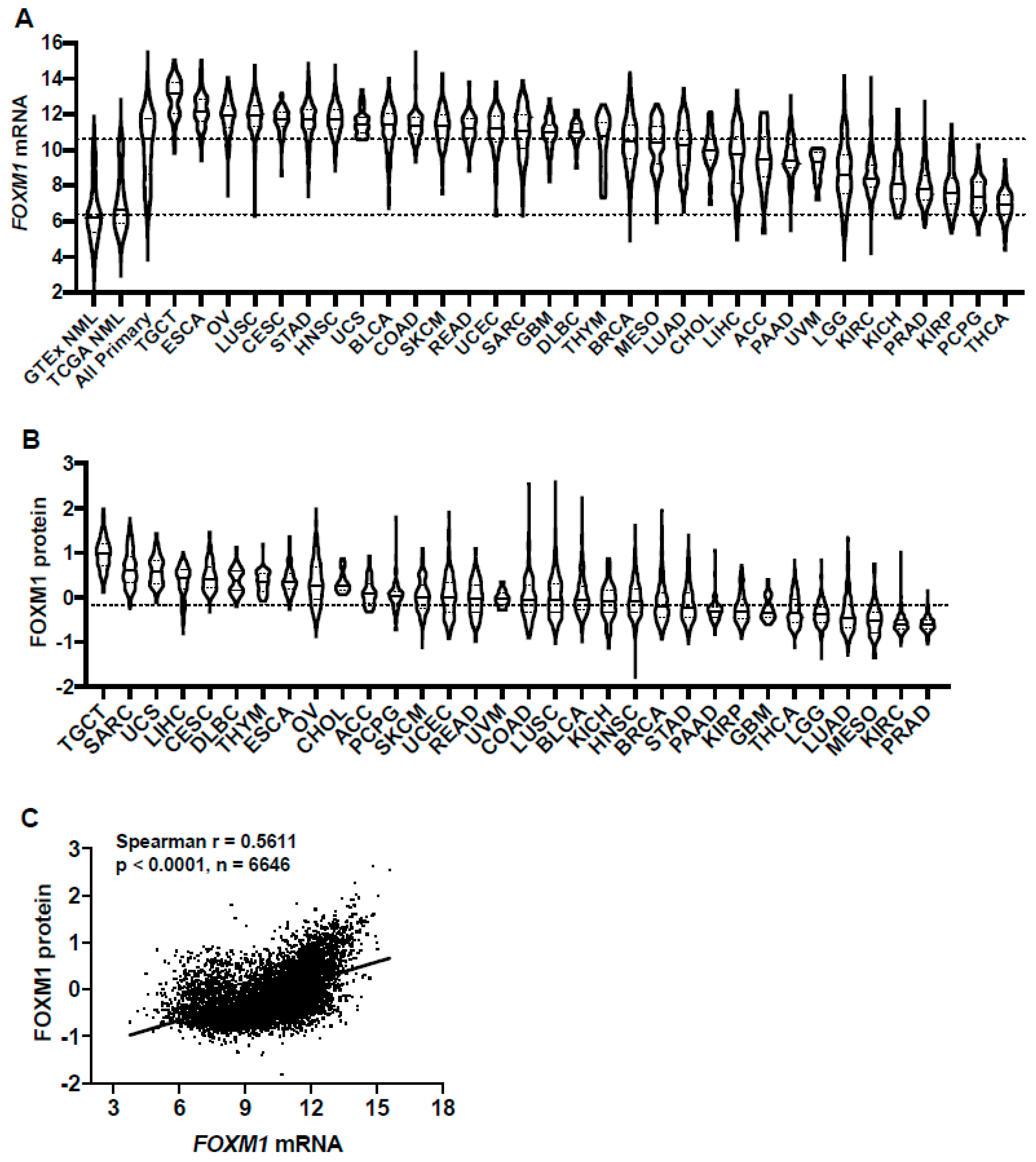
2.1. FOXM1 Expression in Pan-Cancer
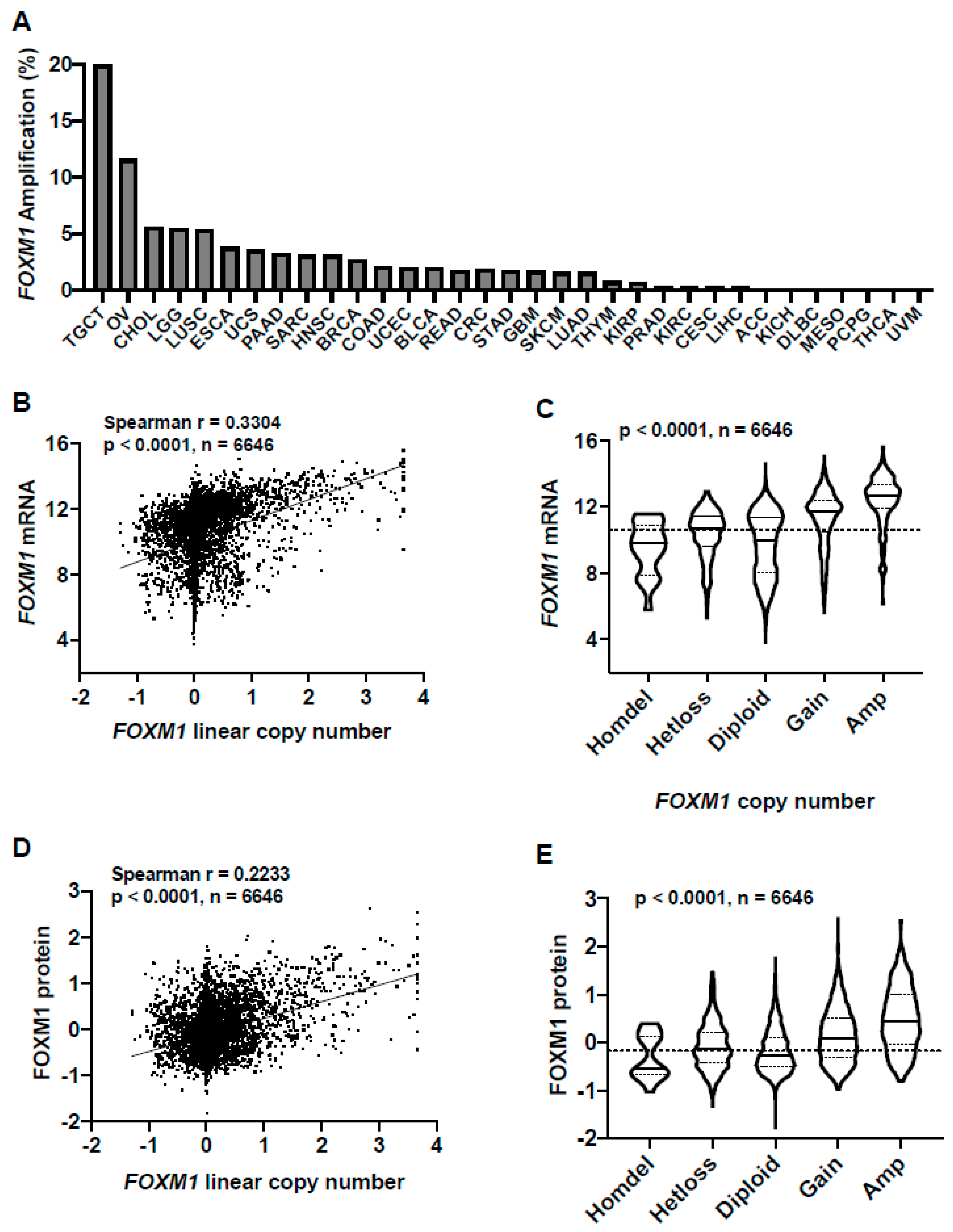
2.2. FOXM1 Overexpression in Pan-Cancer Is Associated with FOXM1 Amplification and Copy Number Gain
2.3. FOXM1 Overexpression in Pan-Cancer Correlates with Genomic Instability
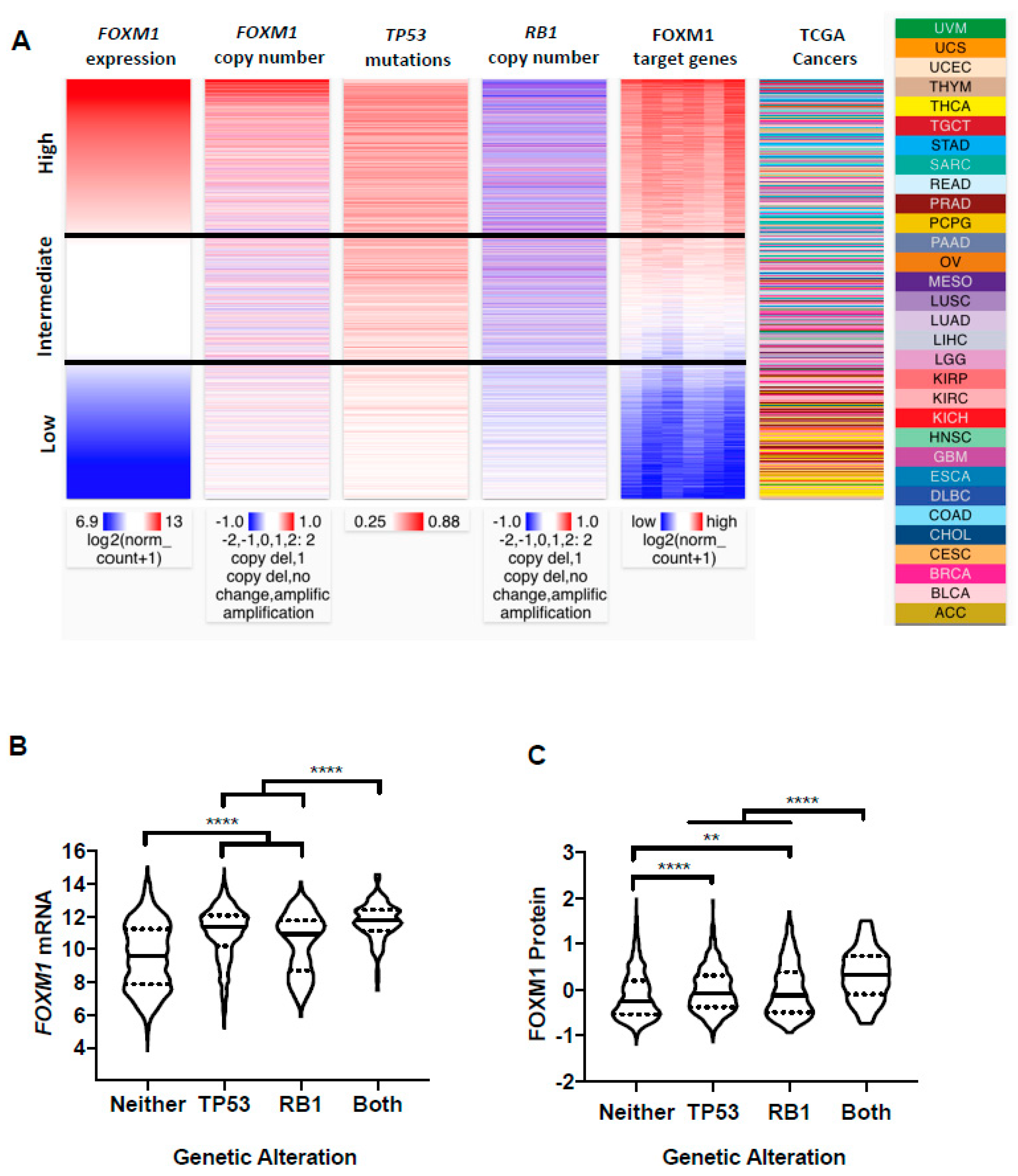
2.4. FOXM1 Overexpression in Pan-Cancer Correlates with TP53 and RB1 Alterations
2.5. High-Grade Serous Ovarian Cancer (HGSC) and Basal Breast Cancer (BBC) Have Highly Similar FOXM1 Alterations
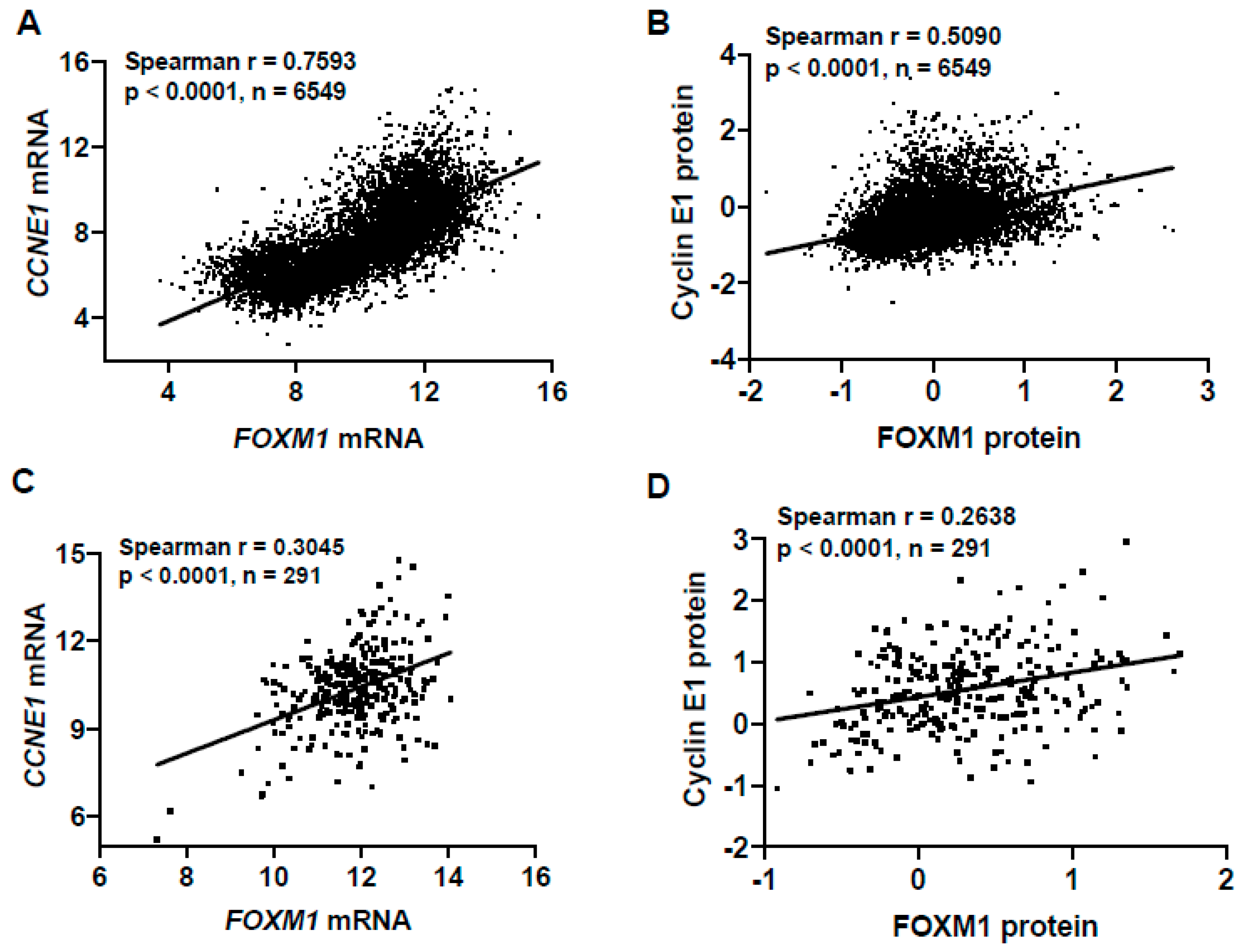
2.6. Deregulation of the Rb-E2F Pathway, Including Cyclin E1 Expression, Promotes FOXM1 Overexpression in Fallopian Tube Epithelial (FTE) Cells, An HGSC Precursor Cell Model
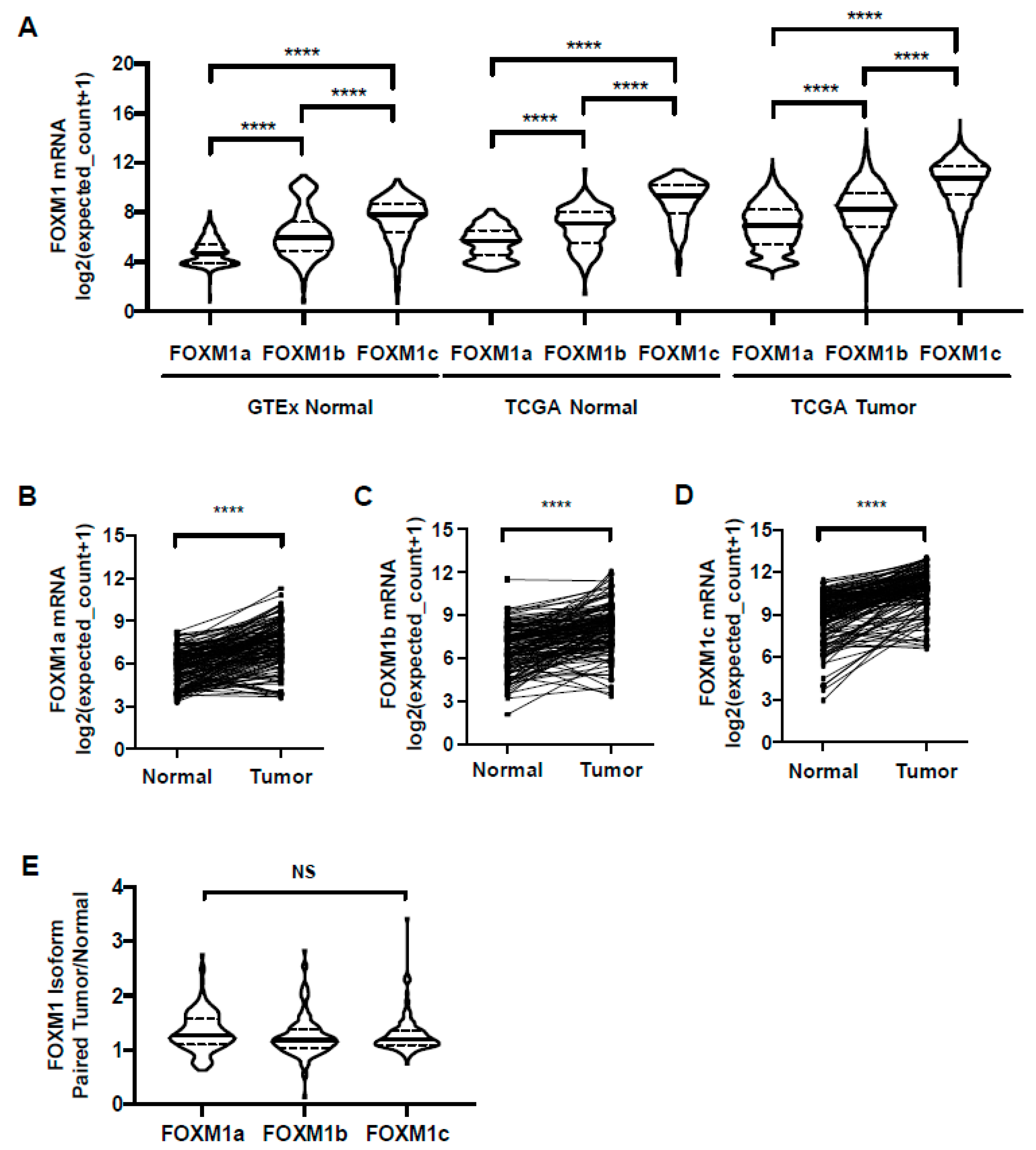
2.7. FOXM1 Isoform Expression in GTEx Normal, TCGA Normal, and TCGA Pan-Cancer Tissues
2.8. FOXM1 Isoforms b and c Have Similar Transcriptional Activity
2.9. Lack of Evidence for A FOXM1 Auto-Regulatory Loop
3. Discussion
4. Materials and Methods
4.1. The Cancer Genome Atlas (TCGA), Cancer Cell Line Encyclopedia (CCLE), Genotype-Tissue Expression (GTEx) Data Sets
4.2. Reverse Transcriptase Quantitative PCR (RT-qPCR)
4.3. Western Blot Analysis
4.4. Cell Culture
4.5. Lentivirus Production and Cell Line Transduction
4.6. Plasmid Constructs
4.7. CRISPR Knockout
4.8. Promoter Activity Luciferase Reporter Assay
4.9. RNA Sequencing (RNA-seq) Analysis
4.10. Statistical Analyses
4.11. Data Deposit
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; Roebuck, K.A.; Costa, R.H. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.H. FoxM1 dances with mitosis. Nat. Cell Biol. 2005, 7, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Wonsey, D.R.; Follettie, M.T. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005, 65, 5181–5189. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Jiang, P.; Freedman, M.L.; Liu, J.S.; Liu, X.S. Inference of transcriptional regulation in cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 7731–7736. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinković, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Kalin, T.V. Is there potential to target FOXM1 for ‘undruggable’ lung cancers? Expert Opin. Ther. Targets 2015, 19, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Bhat, U.G.; Halasi, M.; Gartel, A.L. FoxM1 is a general target for proteasome inhibitors. PLoS ONE 2009, 4, e6593. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. FoxM1 inhibitors as potential anticancer drugs. Expert Opin. Ther. Targets 2008, 12, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. Thiostrepton, proteasome inhibitors and FOXM1. Cell Cycle 2011, 10, 4341–4342. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, L.; Du, Y.; Zheng, H.; Zhang, P.; Sun, Y.; Wang, Y.; Chen, J.; Ding, P.; Wang, N.; et al. A novel FOXM1 isoform, FOXM1D, promotes epithelial-mesenchymal transition and metastasis through ROCKs activation in colorectal cancer. Oncogene 2017, 36, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.L.; DeBoever, C.; Jepsen, K.; Saenz, C.C.; Carson, D.A.; Frazer, K.A. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, E3050–E3057. [Google Scholar] [CrossRef]
- Delpuech, O.; Griffiths, B.; East, P.; Essafi, A.; Lam, E.W.; Burgering, B.; Downward, J.; Schulze, A. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol. Cell. Biol. 2007, 27, 4917–4930. [Google Scholar] [CrossRef]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef]
- Lam, A.K.; Ngan, A.W.; Leung, M.H.; Kwok, D.C.; Liu, V.W.; Chan, D.W.; Leung, W.Y.; Yao, K.M. FOXM1b, which is present at elevated levels in cancer cells, has a greater transforming potential than FOXM1c. Front. Oncol. 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dai, B.; Kang, S.H.; Ban, K.; Huang, F.J.; Lang, F.F.; Aldape, K.D.; Xie, T.X.; Pelloski, C.E.; Xie, K.; et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.Y.; Tong, T.H.; Leung, W.Y.; Yao, K.M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1. Methods Mol. Biol. 2010, 647, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.T.; Wong, S.T.; Neill, G.W.; Ghali, L.R.; Philpott, M.P.; Quinn, A.G. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002, 62, 4773–4780. [Google Scholar] [PubMed]
- Barger, C.J.; Zhang, W.; Hillman, J.; Stablewski, A.B.; Higgins, M.J.; Vanderhyden, B.C.; Odunsi, K.; Karpf, A.R. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget 2015, 6, 27613–27627. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Li, L.; Li, Z.; Le, X.; Huang, C.; Jia, Z.; Cui, J.; Huang, S.; Wang, L.; Xie, K. Dysregulated expression of FOXM1 isoforms drives progression of pancreatic cancer. Cancer Res. 2013, 73, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef]
- Tassi, R.A.; Todeschini, P.; Siegel, E.R.; Calza, S.; Cappella, P.; Ardighieri, L.; Cadei, M.; Bugatti, M.; Romani, C.; Bandiera, E.; et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res. 2017, 36, 63. [Google Scholar] [CrossRef]
- Ma, R.Y.; Tong, T.H.; Cheung, A.M.; Tsang, A.C.; Leung, W.Y.; Yao, K.M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005, 118, 795–806. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. Despite its strong transactivation domain, transcription factor FOXM1c is kept almost inactive by two different inhibitory domains. Biol. Chem. 2006, 387, 963–976. [Google Scholar] [CrossRef]
- Halasi, M.; Gartel, A.L. FOX(M1) news—It is cancer. Mol. Cancer Ther. 2013, 12, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Delahaye-Sourdeix, M.; Oliver, J.; Timofeeva, M.N.; Gaborieau, V.; Johansson, M.; Chabrier, A.; Wozniak, M.B.; Brenner, D.R.; Vallee, M.P.; Anantharaman, D.; et al. The 12p13.33/RAD52 Locus and Genetic Susceptibility to Squamous Cell Cancers of Upper Aerodigestive Tract. PLoS ONE 2015, 10, e0117639. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Aya-Bonilla, C.; Gandhi, M.K.; Lea, R.A.; Wellwood, J.; Wood, P.; Marlton, P.; Griffiths, L.R. Integrative genomic profiling reveals conserved genetic mechanisms for tumorigenesis in common entities of non-Hodgkin’s lymphoma. Genes Chromosomes Cancer 2011, 50, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Deshmukh, H.; Payton, J.E.; Dunham, C.; Scheithauer, B.W.; Tihan, T.; Prayson, R.A.; Guha, A.; Bridge, J.A.; Ferner, R.E.; et al. Array-based comparative genomic hybridization identifies CDK4 and FOXM1 alterations as independent predictors of survival in malignant peripheral nerve sheath tumor. Clin. Cancer Res. 2011, 17, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Buccitelli, C.; Salgueiro, L.; Rowald, K.; Sotillo, R.; Mardin, B.R.; Korbel, J.O. Pan-cancer analysis distinguishes transcriptional changes of aneuploidy from proliferation. Genome Res. 2017, 27, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef]
- Ottesen, A.M.; Skakkebaek, N.E.; Lundsteen, C.; Leffers, H.; Larsen, J.; Rajpert-De Meyts, E. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer 2003, 38, 117–125. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. (University of Nebraska Medical Center, Omaha, NE, USA). Personal communication, 2019.
- Pfister, K.; Pipka, J.L.; Chiang, C.; Liu, Y.; Clark, R.A.; Keller, R.; Skoglund, P.; Guertin, M.J.; Hall, I.M.; Stukenberg, P.T. Identification of Drivers of Aneuploidy in Breast Tumors. Cell Rep. 2018, 23, 2758–2769. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Halasi, M.; Gartel, A.L. p53 negatively regulates expression of FoxM1. Cell Cycle 2009, 8, 3425–3427. [Google Scholar] [CrossRef] [PubMed]
- Millour, J.; de Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Drapkin, R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat. Protoc. 2012, 7, 1755–1764. [Google Scholar] [CrossRef]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschöp, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Heinen, K.; Weghuis, D.O.; de Bruijn, D.; van Kessel, A.G.; Clevers, H. The human TRIDENT/HFH-11/FKHL16 gene: Structure, localization, and promoter characterization. Genomics 1997, 46, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Wilson, A.; Clevers, H. The winged-helix transcription factor Trident is expressed in actively dividing lymphocytes. Immunobiology 1997, 198, 157–161. [Google Scholar] [CrossRef]
- Halasi, M.; Gartel, A.L. A novel mode of FoxM1 regulation: Positive auto-regulatory loop. Cell Cycle 2009, 8, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.H.; Black, M.; Ustiyan, V.; Le, T.; Fulford, L.; Sridharan, A.; Medvedovic, M.; Kalinichenko, V.V.; Whitsett, J.A.; Kalin, T.V. SPDEF inhibits prostate carcinogenesis by disrupting a positive feedback loop in regulation of the Foxm1 oncogene. PLoS Genet. 2014, 10, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Q.; Xie, Y.; Qiao, X.; Zhang, S.; Wang, Y.; Yang, Y.; Zhang, B. Identification of FOXM1 as a specific marker for triple-negative breast cancer. Int. J. Oncol. 2019, 54, 87–97. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef]
- Levanon, K.; Sapoznik, S.; Bahar-Shany, K.; Brand, H.; Shapira-Frommer, R.; Korach, J.; Hirsch, M.S.; Roh, M.H.; Miron, A.; Liu, J.F.; et al. FOXO3a loss is a frequent early event in high-grade pelvic serous carcinogenesis. Oncogene 2014, 33, 4424–4432. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef]
- Molinuevo, R.; Freije, A.; de Pedro, I.; Stoll, S.W.; Elder, J.T.; Gandarillas, A. FOXM1 allows human keratinocytes to bypass the oncogene-induced differentiation checkpoint in response to gain of MYC or loss of p53. Oncogene 2017, 36, 956–965. [Google Scholar] [CrossRef]
- Schimmel, J.; Eifler, K.; Sigurðsson, J.O.; Cuijpers, S.A.; Hendriks, I.A.; Verlaan-de Vries, M.; Kelstrup, C.D.; Francavilla, C.; Medema, R.H.; Olsen, J.V.; et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell 2014, 53, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.; Medema, R.H. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.A.; Gormally, M.V.; Marsico, G.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. FOXM1 binds directly to non-consensus sequences in the human genome. Genome Biol. 2015, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Akbani, R.; Zhao, W.; Lu, Y.; Weinstein, J.N.; Mills, G.B.; Liang, H. Explore, Visualize, and Analyze Functional Cancer Proteomic Data Using the Cancer Proteome Atlas. Cancer Res. 2017, 77, e51–e54. [Google Scholar] [CrossRef]
- Akbani, R.; Ng, P.K.; Werner, H.M.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.Y.; Yoshihara, K.; Li, J.; et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef]
- Safikhani, Z.; Smirnov, P.; Thu, K.L.; Silvester, J.; El-Hachem, N.; Quevedo, R.; Lupien, M.; Mak, T.W.; Cescon, D.; Haibe-Kains, B. Gene isoforms as expression-based biomarkers predictive of drug response in vitro. Nat. Commun. 2017, 8, 1126. [Google Scholar] [CrossRef]
- Petrovic, V.; Costa, R.H.; Lau, L.F.; Raychaudhuri, P.; Tyner, A.L. Negative regulation of the oncogenic transcription factor FoxM1 by thiazolidinediones and mithramycin. Cancer Biol. Ther. 2010, 9, 1008–1016. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. https://doi.org/10.3390/cancers11020251
Barger CJ, Branick C, Chee L, Karpf AR. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers. 2019; 11(2):251. https://doi.org/10.3390/cancers11020251
Chicago/Turabian StyleBarger, Carter J, Connor Branick, Linda Chee, and Adam R. Karpf. 2019. "Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer" Cancers 11, no. 2: 251. https://doi.org/10.3390/cancers11020251
APA StyleBarger, C. J., Branick, C., Chee, L., & Karpf, A. R. (2019). Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers, 11(2), 251. https://doi.org/10.3390/cancers11020251