Paclitaxel-Induced Src Activation Is Inhibited by Dasatinib Treatment, Independently of Cancer Stem Cell Properties, in a Mouse Model of Ovarian Cancer


 ,
, 
Abstract
1. Introduction
2. Results
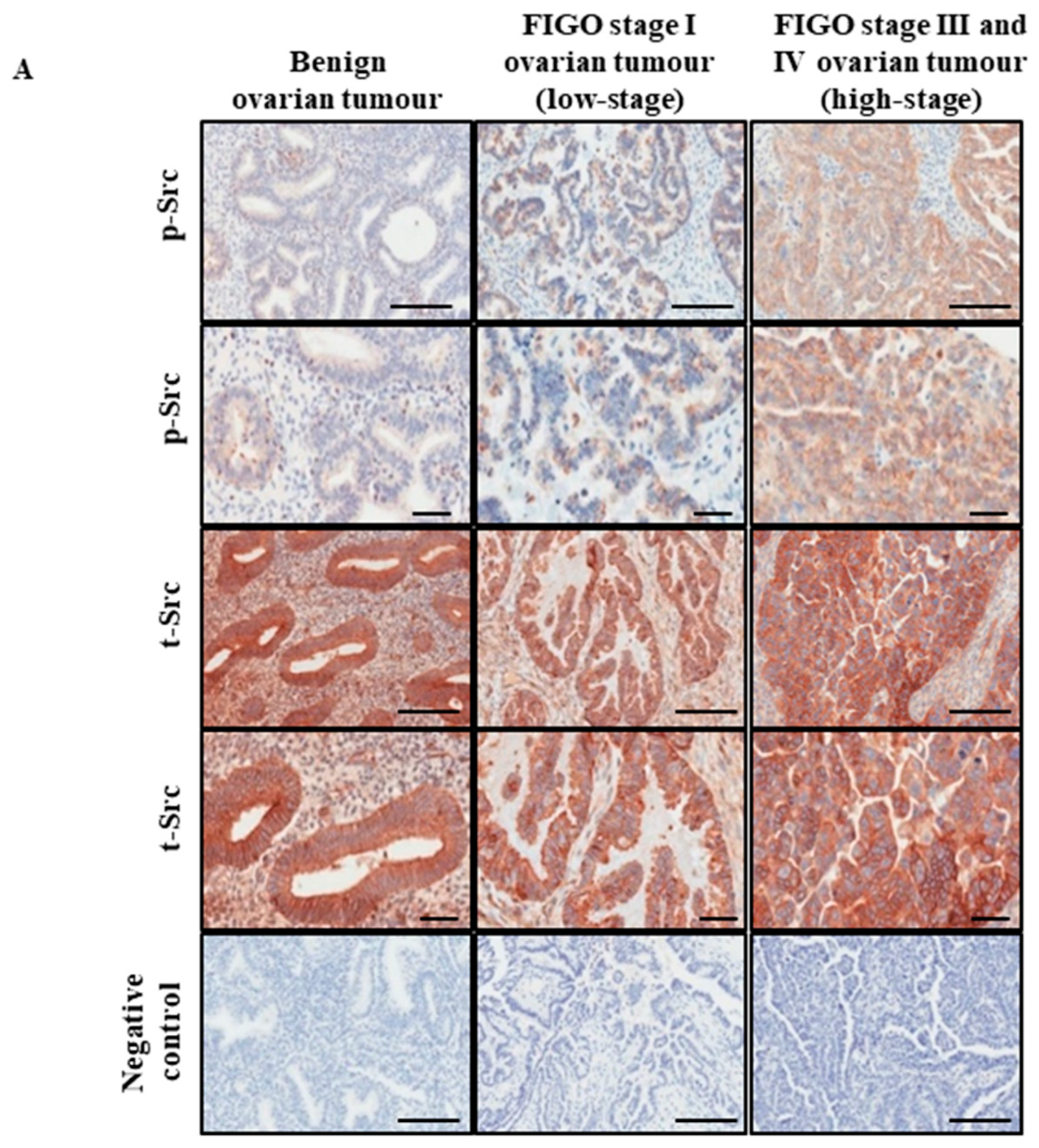
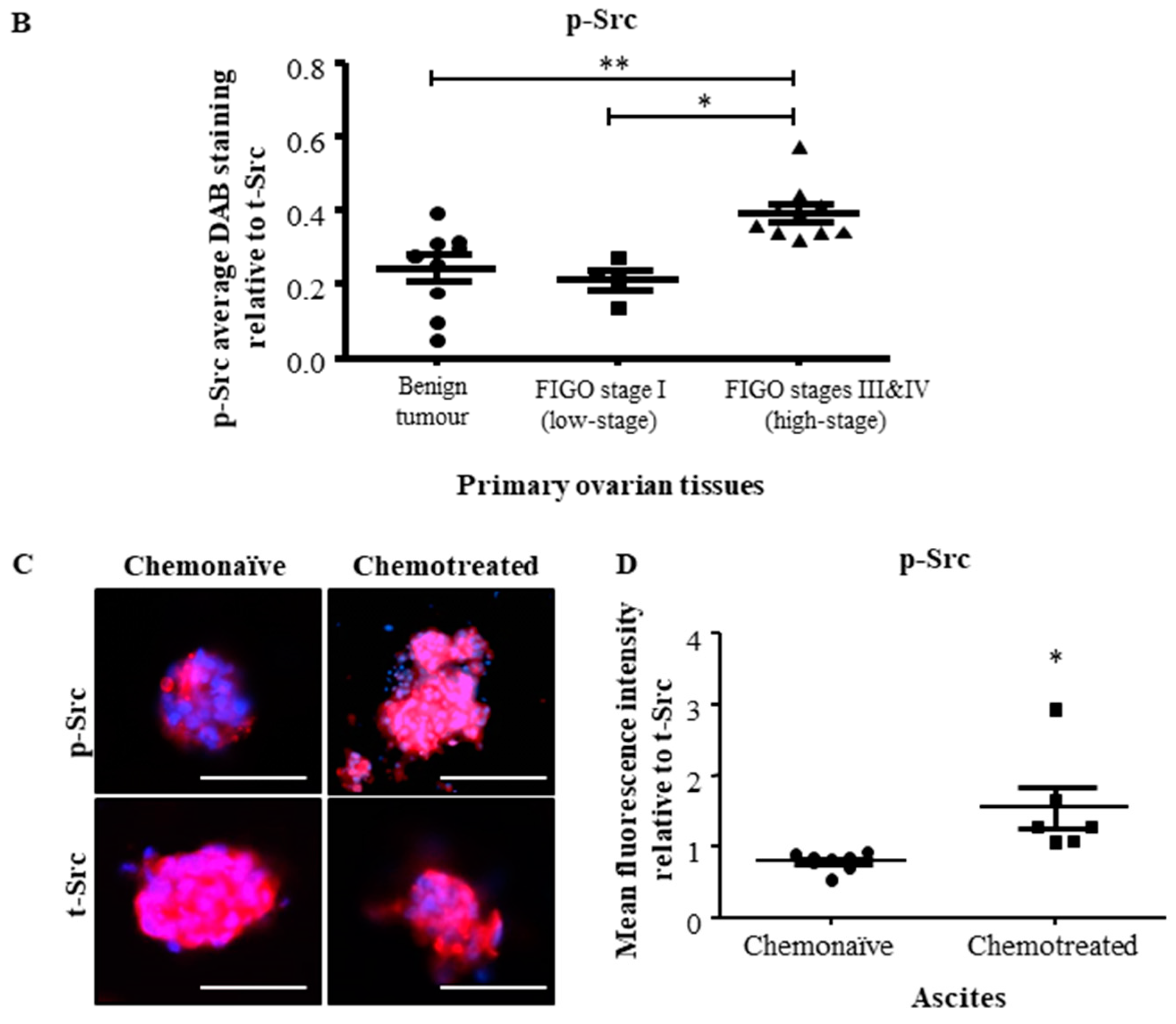
2.1. Active Src Levels Are Increased in the Primary Ovarian Tumours of High-Stage Serous Ovarian Cancer Patients and in the Ascites-Derived Tumour Cells of Advanced-Stage Recurrent Patients
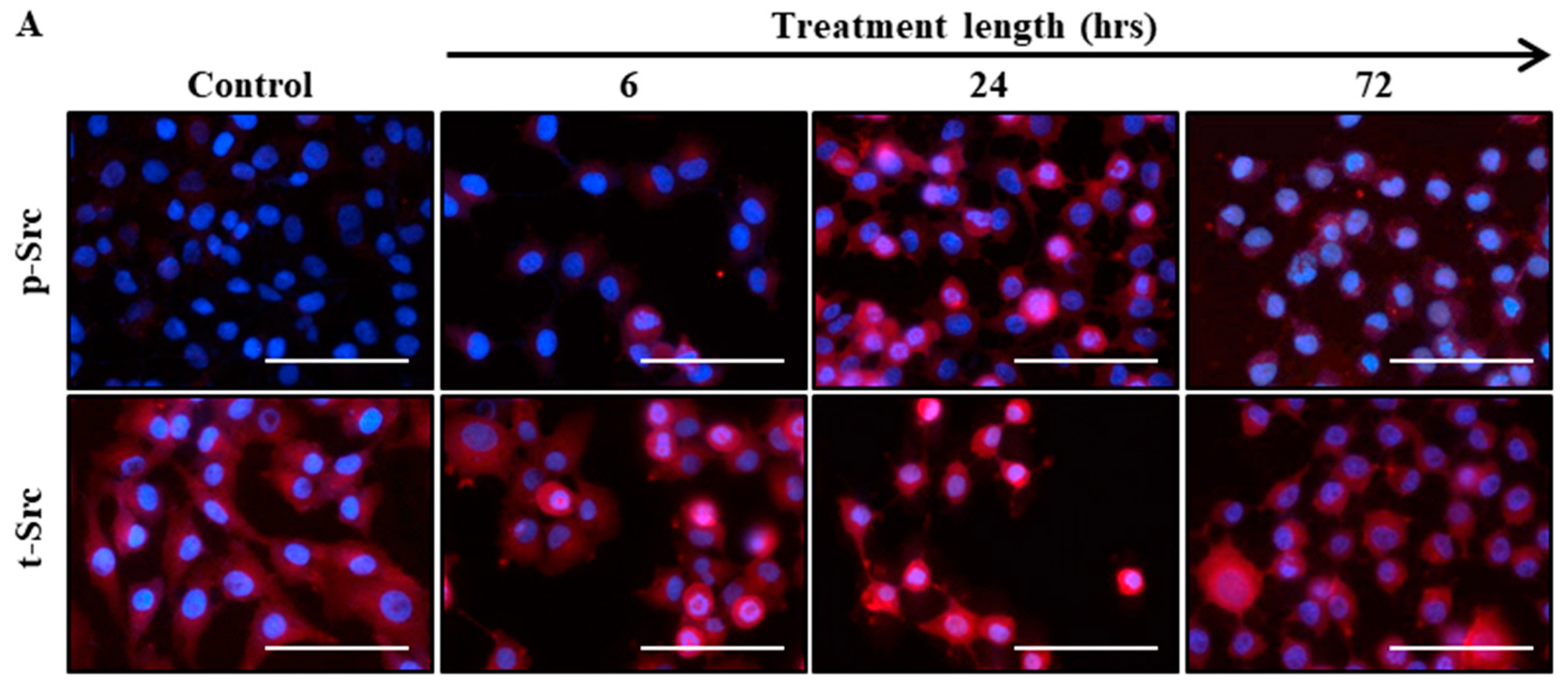
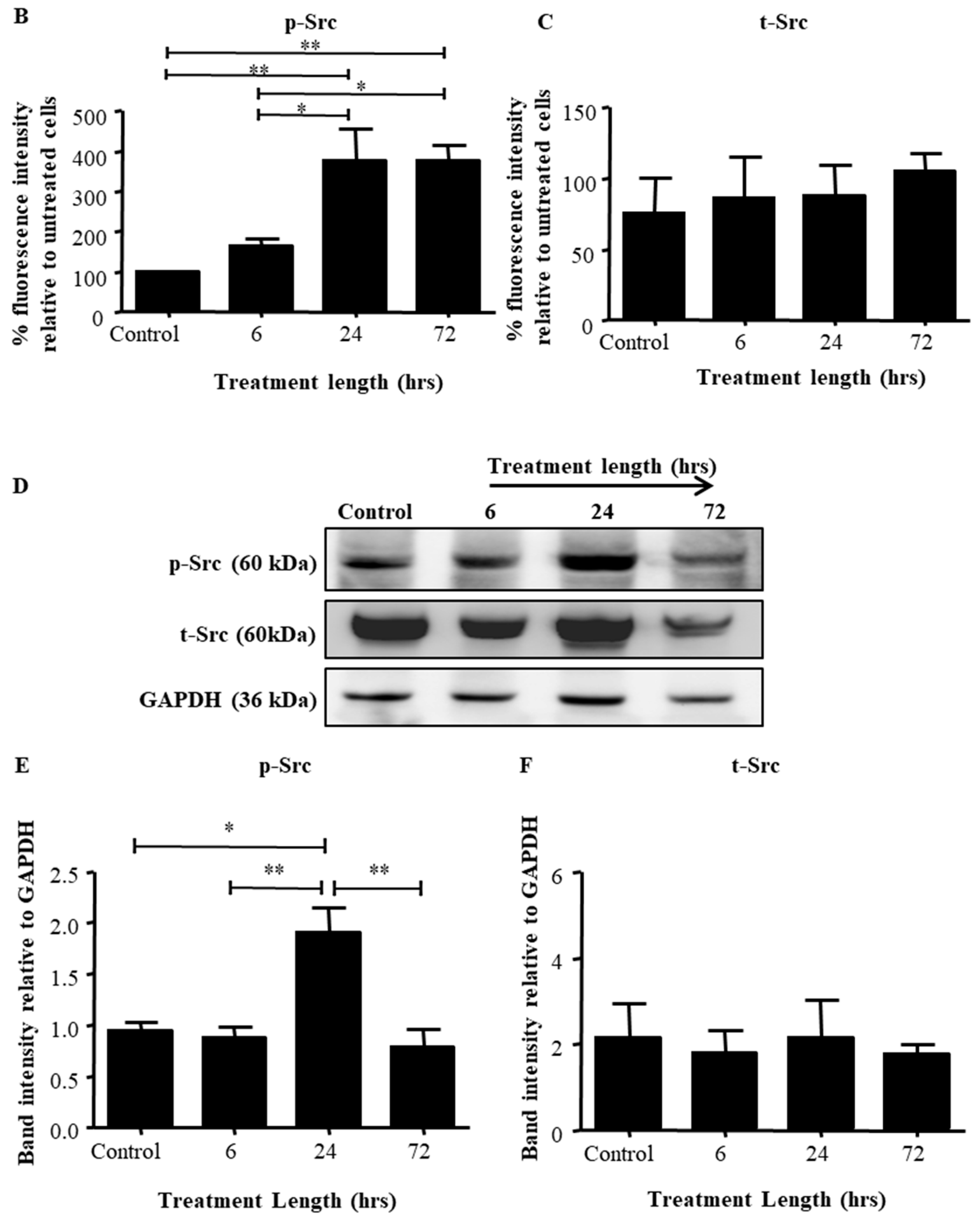
2.2. Src Activation Was Transiently Induced in Ovarian Cancer Cell Lines by Paclitaxel Treatment
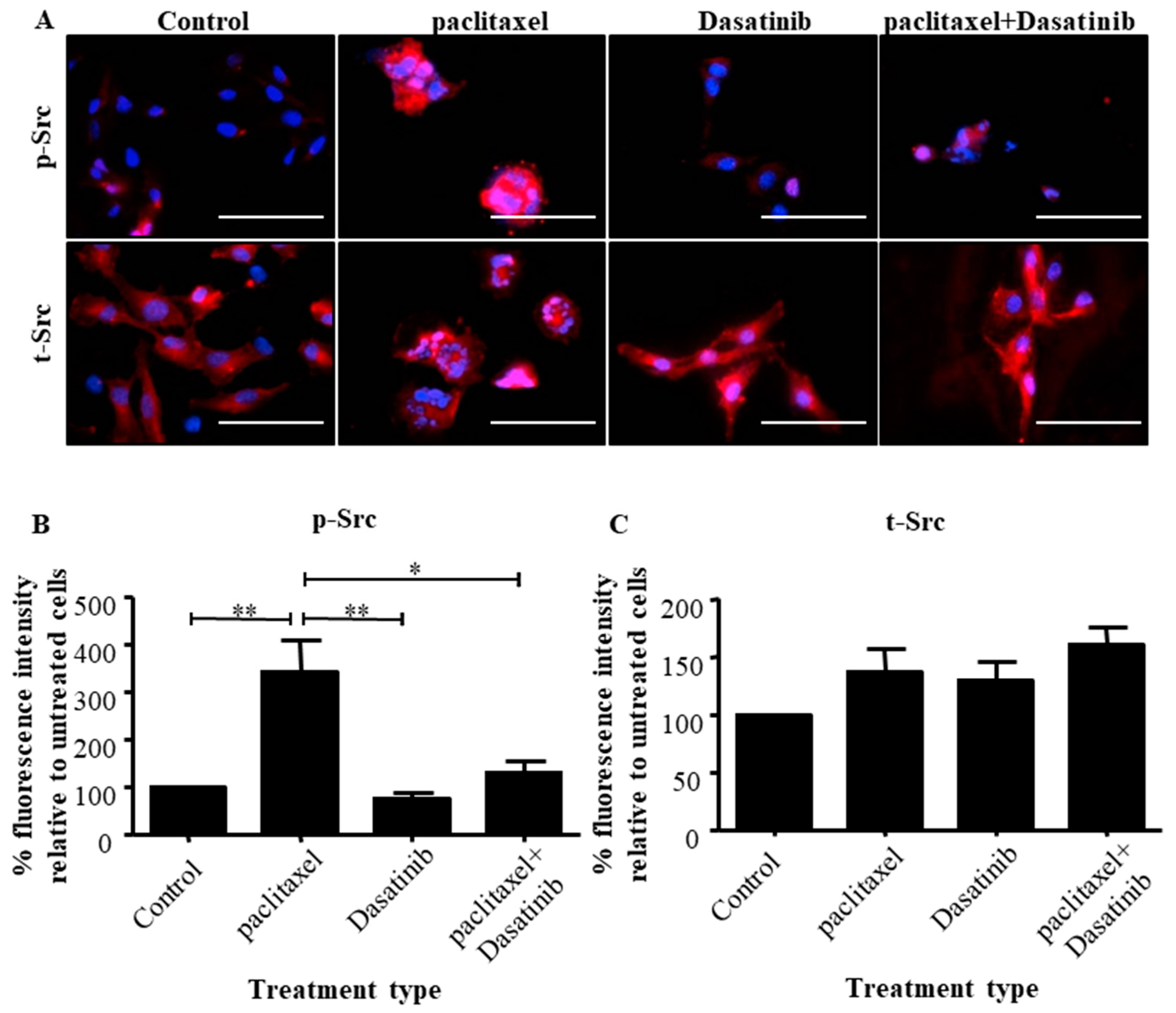
2.3. The Addition of Dasatinib Suppressed Paclitaxel-Induced Src Activation in Ovarian Cancer Cells
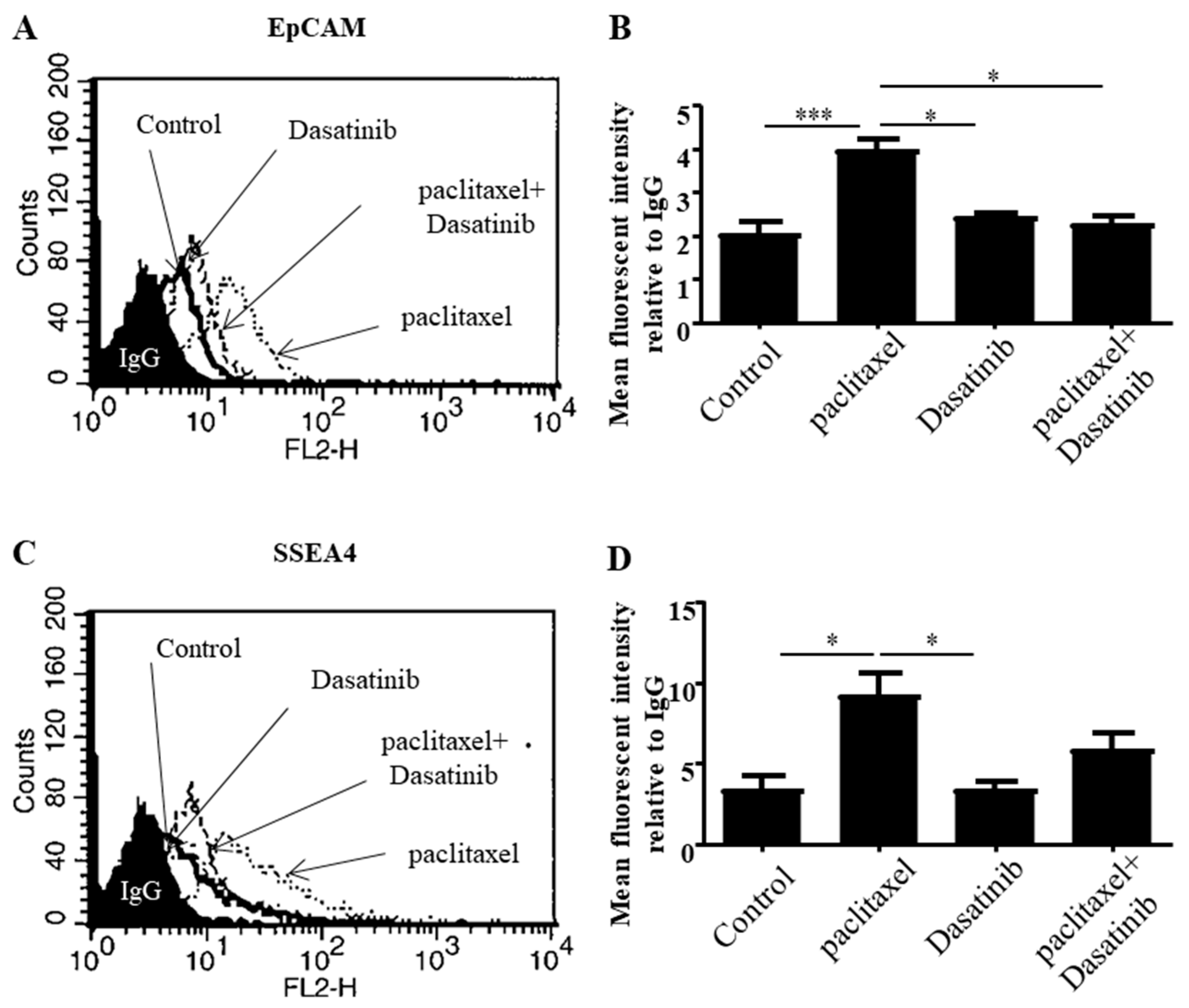
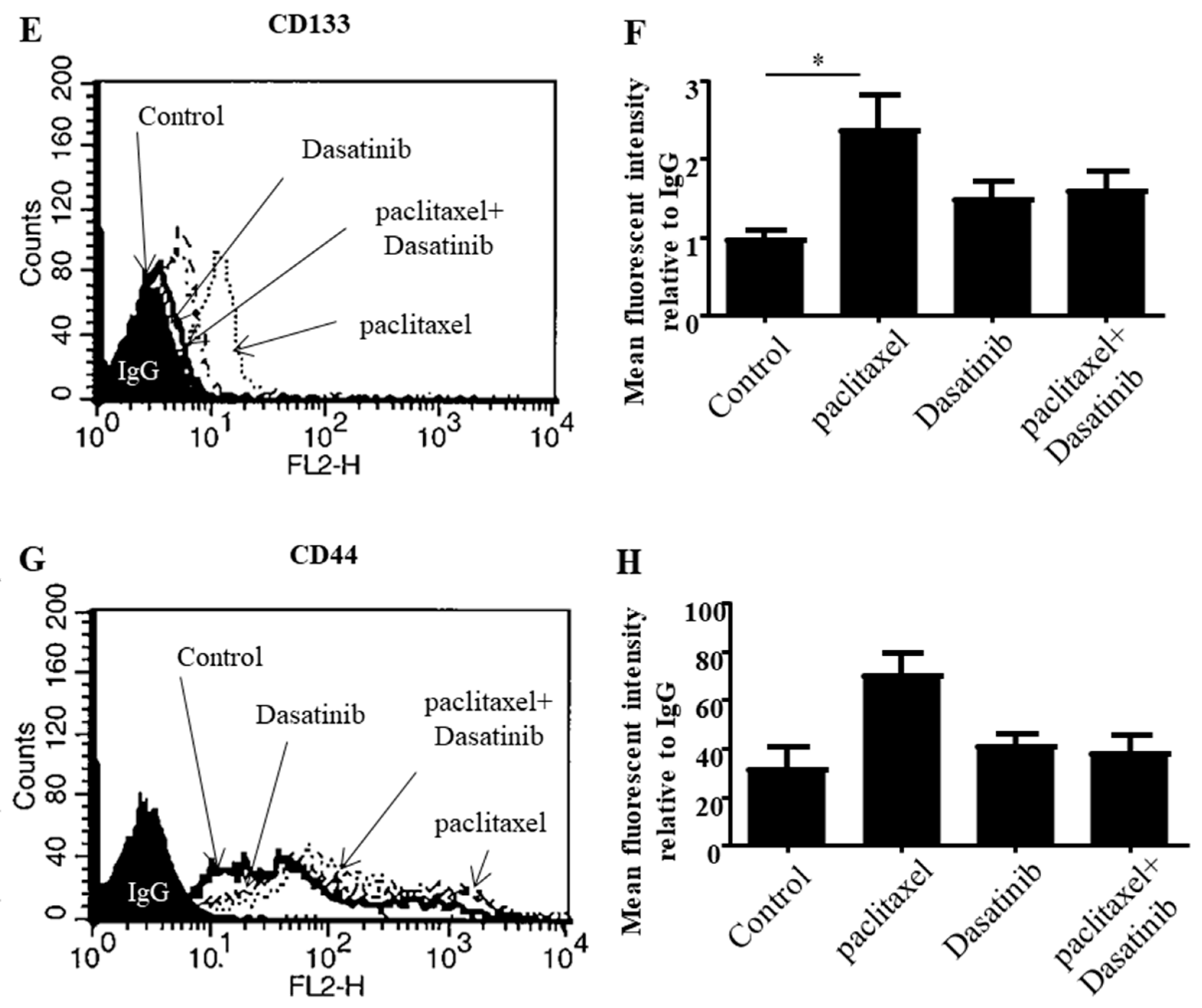
2.4. Dasatinib Reduced Some Paclitaxel-Induced CSC-Like Marker Expression in Ovarian Cancer Cells
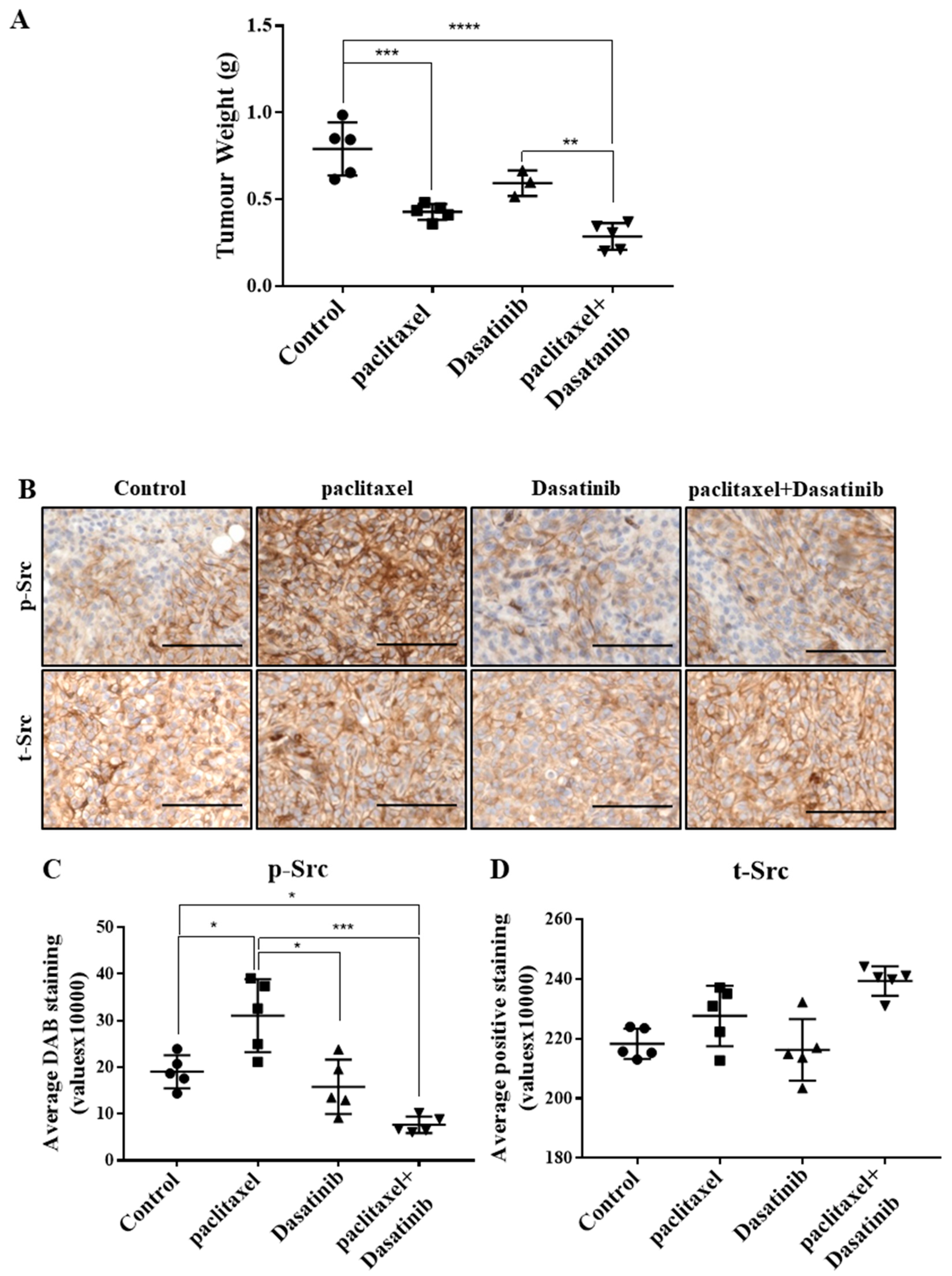
2.5. Combination Treatment Had no Significant Effect in Reducing Tumour Burden Over Paclitaxel but Inhibited Src Activation in HEY Derived Xenografts
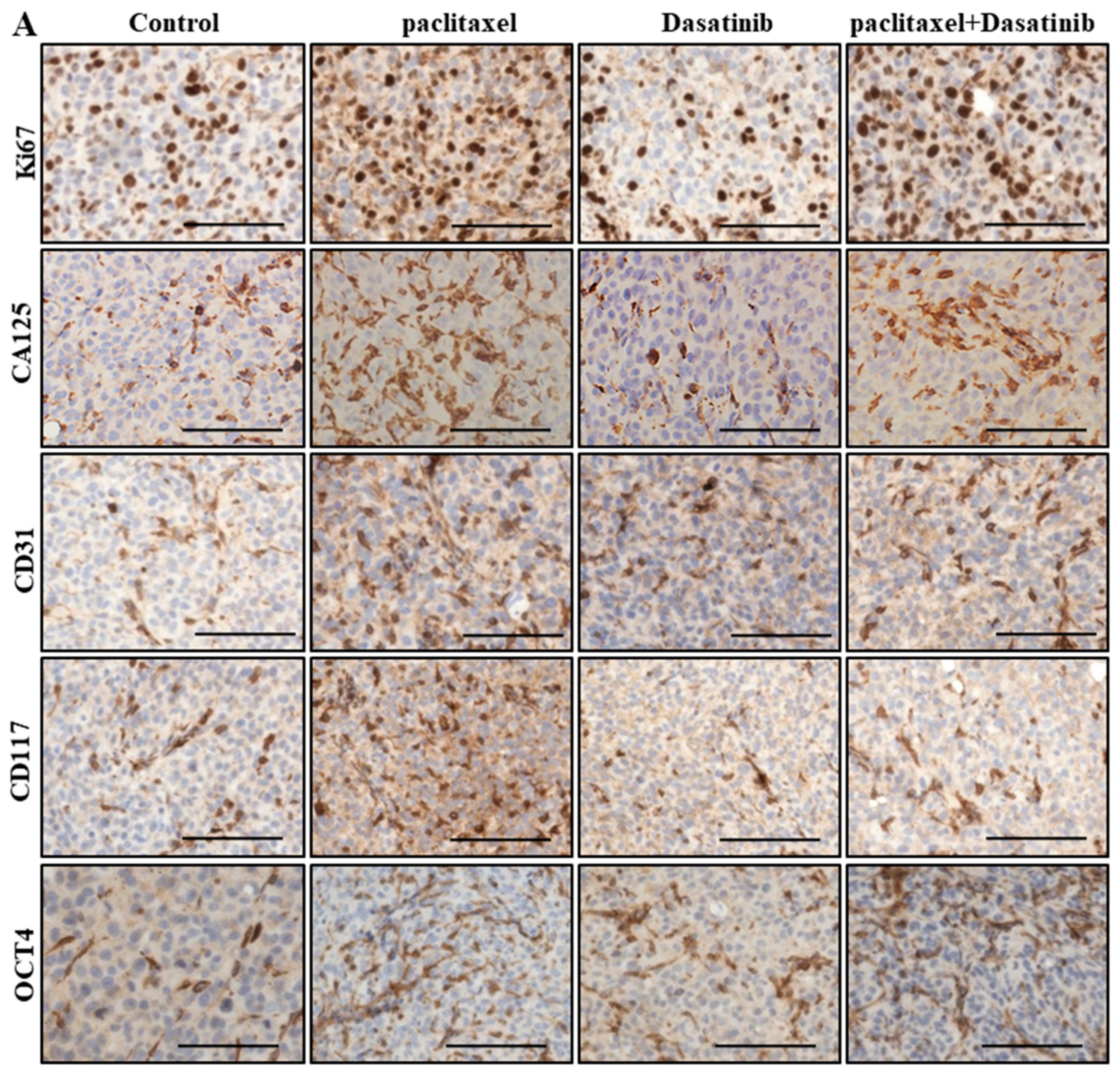
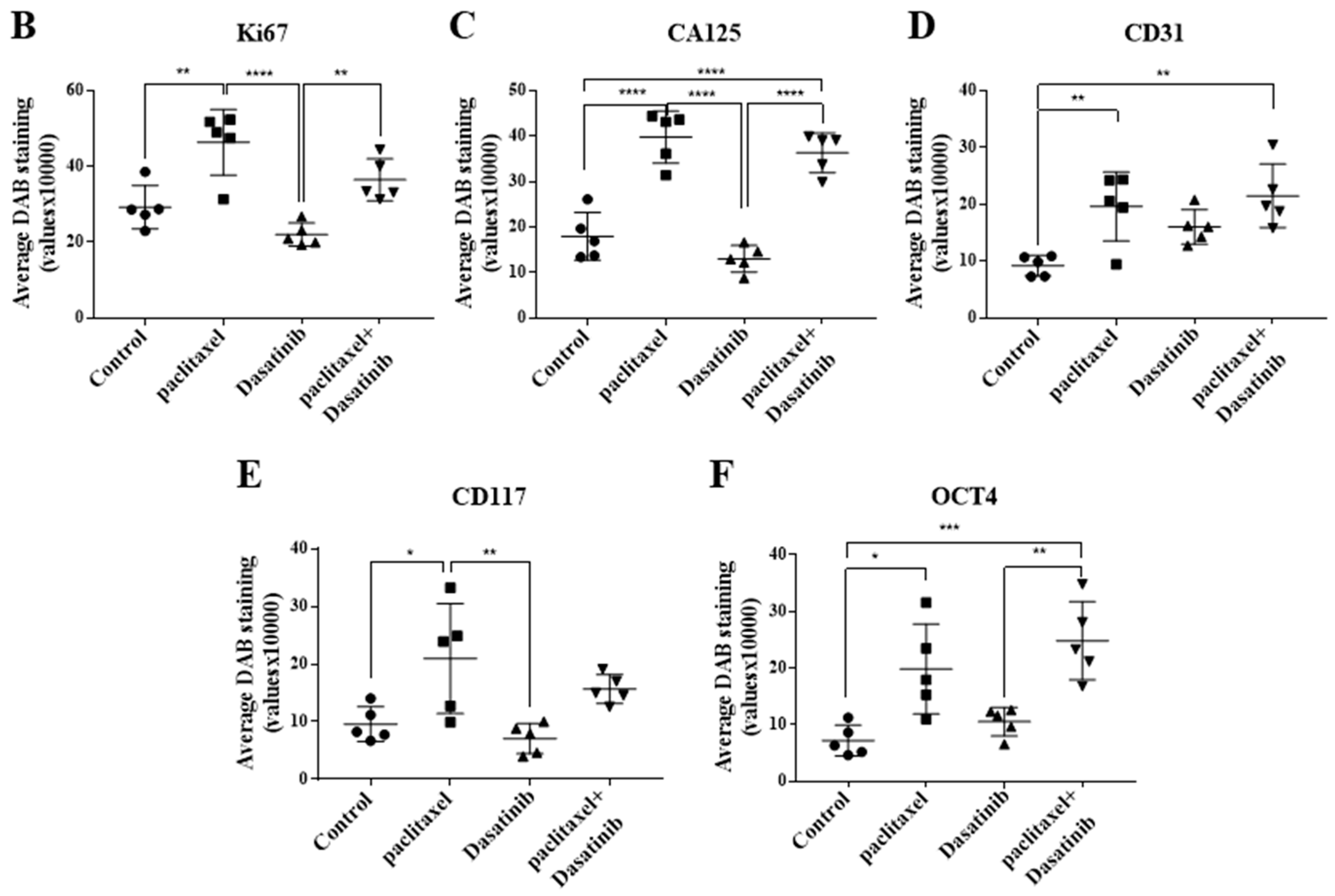
2.6. Inhibition of p-Src by Dasatinib Had no Significant Effect on Paclitaxel-Induced Expression of CSC, Proliferation, Tumourigenicity or the Vascularization Markers
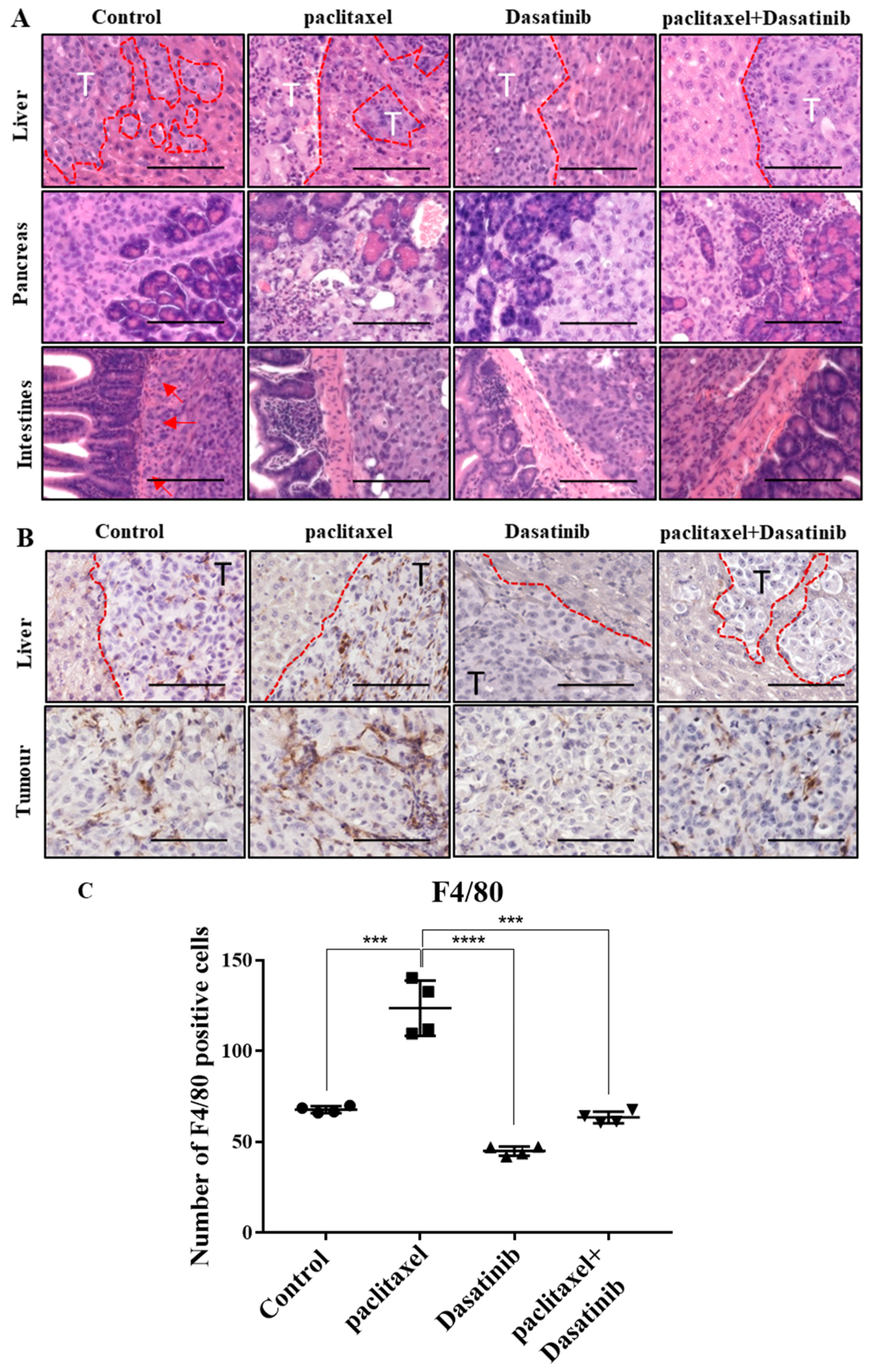
2.7. Tumours Treated with Dasatinib in Combination with Paclitaxel Had a Reduced Tendency to Invade Peritoneal Organs and Had Significantly Reduced Mononuclear Infiltrate
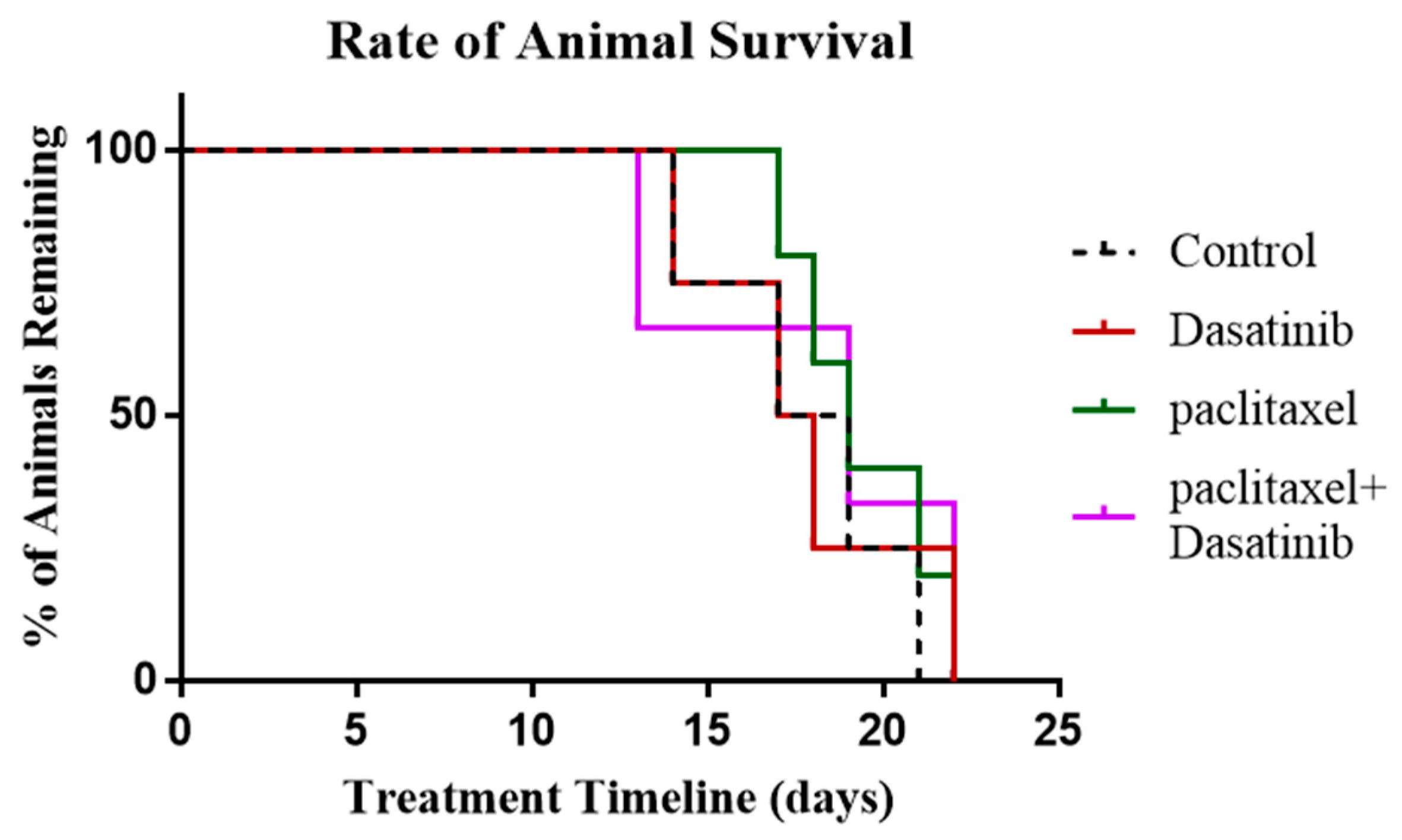
2.8. Termination of Paclitaxel, Dasatinib or a Combination of Both Treatments Resulted in Robust Tumour Development with no Change in Survival of Tumour Bearing Mice
3. Discussion
4. Materials and Methods
4.1. Human Ethics Approval
4.2. Human Primary Tissue Collection and Preparation
4.3. Human Ascites Collection
4.4. Cell Cultures
4.5. Drug Preparation
4.6. Chemosensitivity Assay
4.7. Antibodies and Reagents
4.8. Cell Preparation for Immunofluorescence
4.9. Western Blotting
4.10. Flow Cytometry
4.11. Mouse Experiments
4.12. Intraperitoneal Ovarian Cancer Model-Treatment Regimen
4.13. Processing of Mice Tissues
4.14. Haematoxylin and Eosin Stain (H&E Stain)
4.15. Immunohistochemical Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F. Treatment goals in ovarian cancer. Int. J. Gynecol. Cancer 2005, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Felipe De Sousa, E.M.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann Oncol. 2012, 23, 2605–2612. [Google Scholar]
- Sehouli, J.; Senyuva, F.; Fotopoulou, C.; Neumann, U.; Denkert, C.; Werner, L.; Gülten, O.Ö. Intra-abdominal tumor dissemination pattern and surgical outcome in 214 patients with primary ovarian cancer. J. Surg. Oncol. 2009, 99, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, W.; Fan, C.; Chen, X.; Lu, R.; Liu, Y.; Li, Y.; Shang, Y.; Yin, D.; Zhang, S.; Huang, Q.; et al. Endothelium originated from colorectal cancer stem cells constitute cancer blood vessels. Cancer Sci. 2017, 108, 1357–1367. [Google Scholar] [CrossRef]
- Vermeulen, L.; Felipe De Sousa, E.M.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Aguilar-Gallardo, C.; Rutledge, E.C.; Martinez-Arroyo, A.M.; Hidalgo, J.J.; Domingo, S.; Simon, C. Overcoming challenges of ovarian cancer stem cells: Novel therapeutic approaches. Stem Cell Rev. 2012, 8, 994–1010. [Google Scholar] [CrossRef]
- McCarthy, N. Signalling: SRC and survival. Nat. Rev. Cancer 2012, 12, 80–81. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Chen, C.; Xu, M.M.; Li, J.D.; Xiao, J.; Zhu, X.F. Expression of c-Src and phospho-Src in epithelial ovarian carcinoma. Mol. Cell. Biochem. 2013, 376, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Xu, M.; Hou, T.; Huang, Y.; Yang, C.; Li, J. Dasatinib enhances antitumor activity of paclitaxel in ovarian cancer through Src signaling. Mol. Med. Rep. 2015, 12, 3249–3256. [Google Scholar] [CrossRef]
- Brave, M.; Goodman, V.; Kaminskas, E.; Farrell, A.; Timmer, W.; Pope, S.; Harapanhalli, R.; Saber, H.; Morse, D.; Bullock, J.; et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin. Cancer Res. 2008, 14, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, D.G.; Tsimberidou, A.M. Dasatinib in chronic myeloid leukemia: A review. Ther. Clin. Risk Manag. 2009, 5, 281–289. [Google Scholar] [PubMed]
- Konecny, G.E.; Glas, R.; Dering, J.; Manivong, K.; Qi, J.; Finn, R.S.; Yang, G.R.; Hong, K.L.; Ginther, C.; Winterhoff, B.; et al. Activity of the multikinase inhibitor Dasatinib against ovarian cancer cells. Br. J. Cancer 2009, 101, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Teoh, D.; Ayeni, T.A.; Rubatt, J.M.; Adams, D.J.; Grace, L.; Starr, M.D.; Barry, W.T.; Berchuck, A.; Murphy, S.K.; Secord, A.A.; et al. Dasatinib (BMS-35482) has synergistic activity with paclitaxel and carboplatin in ovarian cancer cells. Gynecol. Oncol. 2011, 121, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Luwor, R.; Burns, C.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 2018, 9, 16599–16618. [Google Scholar] [CrossRef]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Cancerous ovarian stem cells: Obscure targets for therapy but relevant to chemoresistance. J. Cell. Biochem. 2013, 114, 21–34. [Google Scholar] [CrossRef]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Asp. Med. 2014, 39, 110–125. [Google Scholar] [CrossRef]
- Samardzija, C.; Luwor, R.B.; Volchek, M.; Quinn, M.A.; Findlay, J.K.; Ahmed, N. A critical role of Oct4A in mediating metastasis and disease-free survival in a mouse model of ovarian cancer. Mol. Cancer 2015, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, W.; Yang, X.; Wheeler, C.G.; Langford, C.P.; Wu, L.; Filippova, N.; Friedman, G.K.; Ding, Q.; Fathallah-Shaykh, H.M.; et al. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 2014, 45, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Thibault, B.; Jean-Claude, B. Dasatinib + Gefitinib, a non platinum-based combination with enhanced growth inhibitory, anti-migratory and anti-invasive potency against human ovarian cancer cells. J. Ovarian Res. 2017, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Nakachi, I.; Helfrich, B.A.; Spillman, M.A.; Mickler, E.A.; Olson, C.J.; Rice, J.L.; Coldren, C.D.; Heasley, L.E.; Geraci, M.W.; Stearman, R.S.; et al. PTTG1 Levels Are Predictive of Saracatinib Sensitivity in Ovarian Cancer Cell Lines. Clin. Transl. Sci. 2016, 9, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar] [PubMed]
- Wang, Y.; Li, H. Identification of proteins associated with paclitaxel resistance of epithelial ovarian cancer using iTRAQ-based proteomics. Oncol. Lett. 2018, 15, 9793–9801. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef]
- Kwon, M.J.; Shin, Y.K. Regulation of ovarian cancer stem cells or tumor-initiating cells. Int. J. Mol. Sci. 2013, 14, 6624–6648. [Google Scholar] [CrossRef]
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef]
- Demetri, G.D.; Russo, P.L.; MacPherson, I.R.; Wang, D.; Morgan, J.A.; Brunton, V.G.; Paliwal, P.; Agrawal, S.; Voi, M.; Evans, T.J.; et al. Phase I dose-escalation and pharmacokinetic study of Dasatinib in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 6232–6240. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediat. Inflamm. 2017, 2017, 9294018. [Google Scholar] [CrossRef]
- Avila, N.A.; Dwyer, A.J.; Moss, J. Active Surveillance of Nonfatty Renal Masses in Patients with Lymphangioleiomyomatosis: Use of CT Features and Patterns of Growth to Differentiate Angiomyolipoma From Renal Cancer. AJR Am. J. Roentgenol. 2017, 209, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef]
- Mazzi, P.; Caveggion, E.; Lapinet-Vera, J.A.; Lowell, C.A.; Berton, G. The Src-Family Kinases Hck and Fgr Regulate Early Lipopolysaccharide-Induced Myeloid Cell Recruitment into the Lung and Their Ability to Secrete Chemokines. J. Immunol. 2015, 195, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in Non-Small Cell Lung Cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090–26103. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, W.; Okamoto, I.; Yoshida, T.; Okamoto, K.; Takezawa, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Arao, T.; Yanagihara, K.; et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol. Cancer Ther. 2010, 9, 1188–1197. [Google Scholar] [CrossRef]
- Gomes, E.G.; Connelly, S.F.; Summy, J.M. Targeting the yin and the yang: Combined inhibition of the tyrosine kinase c-Src and the tyrosine phosphatase SHP-2 disrupts pancreatic cancer signaling and biology in vitro and tumor formation in vivo. Pancreas 2013, 42, 795–806. [Google Scholar] [CrossRef]
- Tabariès, S.; Annis, M.G.; Hsu, B.E.; Tam, C.E.; Savage, P.; Park, M.; Siegel, P.M. Lyn modulates Claudin-2 expression and is a therapeutic target for breast cancer liver metastasis. Oncotarget 2015, 6, 9476–9487. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.; Ditzel, H.J. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol. Res. 2015, 100, 250–254. [Google Scholar] [CrossRef]
- Kreutzman, A.; Ladell, K.; Koechel, C.; Gostick, E.; Ekblom, M.; Stenke, L.; Melo, T.; Einsele, H.; Porkka, K.; Price, D.A.; et al. Expansion of highly differentiated CD8+ T-cells or NK-cells in patients treated with Dasatinib is associated with cytomegalovirus reactivation. Leukemia 2011, 25, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.L.A.; Chan, E.; Luwor, R.B.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Enhanced activation of STAT3 in ascites-derived recurrent ovarian tumors: Inhibition of cisplatin-induced STAT3 activation reduced tumorigenicity of ovarian cancer by a loss of cancer stem cell-like characteristics. J. Cancer Stem Cell Res. 2015, 3, e1001. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun. 2015, 6, 6139. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Diagnosis | FIGO Stage | Age at Diagnosis | Genetic Background | Pre-Operative CA125 | Ascites at Diagnosis | Survival |
|---|---|---|---|---|---|---|---|
| Normal | |||||||
| 1 | Normal ovarian epithelium | - | BRCA+ | - | No | - | |
| 2 | Normal ovarian epithelium | - | Peutz-Jegher’s syndrome | - | No | - | |
| Benign | |||||||
| 1 | Benign cyst | - | 19 | - | - | No | - |
| 2 | Benign cyst | - | 55 | - | - | No | - |
| 3 | Large multi cysts ovarian mass | - | 64 | Family history of Ca | - | No | - |
| 4 | Benign cyst | - | 58 | Diagnosed with breast cancer | - | No | - |
| 5 | Large ovarian cyst | - | 62 | Unknown | - | No | - |
| 6 | Benign cyst | - | 48 | HNPCC carrier (colorectal cancer) | - | No | - |
| Low-Stage | |||||||
| 1 | Serous cystadenocarcinoma NOS (Gr3) | Ic | 90 | Unknown | 24 | No | 1 month ALC |
| 2 | Serous cystadenocarcinoma NOS | Ia | 36 | Unknown | 138 | No | 2 years 4 months ALC |
| 3 | Serous cystadenocarcinoma (Gr1) | Ic | 31 | Unknown | 177 | No | 4 years 10 months ALC |
| High-Stage | |||||||
| 1 | Papillary serous adenocarcinoma (Gr3) | 55 | - | 3025 | Yes | 2 years 5 months at death | |
| 2 | Serous cystadenocarcinoma NOS (Gr2) | IIIc | 65 | Unknown | 903 | Yes | 1 year 9 months at death |
| 3 | Carcinoma NOS (Gr3) | III | 62 | BRCA+ | 3058 | Yes | 2 years 7 months at death |
| 4 | Serous cystadenocarcinoma NOS (Gr3) | IIIc | 38 | Unknown | 957 | Yes | 2 years 8 months ALC |
| 5 | Serous surface papillary carcinoma (Gr3) | IIIc | 59 | BRCA2+ | 397 | Yes | 4 years 7 months at death |
| 6 | Papillary serous adenocarcinoma (Gr2) | IIIc | 41 | Family history of ovarian cancer | Unknown | Unknown | 8 years 8 months ALC |
| 7 | Serous cystadenocarcinoma NOS (Gr3) | IIIc | 67 | Unknown | Unknown | Yes | 9 months at death |
| 8 | Papillary serous adenocarcinoma (Gr2) | IIIc | 43 | - | 428 | Yes | 3 years 7 months at death |
| 9 | Papillary serous adenocarcinoma (Gr2) | IV | 58 | Family history of uterine cancer | 3187 | Yes | 6 years 10 months at death |
| 10 | Papillary serous adenocarcinoma (Gr3) | IIIc | 53 | Unknown | 1838 | Yes | 1 year 9 months at death |
| Sample Classification | Diagnosis | Grade | FIGO Stage | Treatment | Age at Diagnosis | Survival |
|---|---|---|---|---|---|---|
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IV | None | 76 | 7 months at death |
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IIIc | None | 64 | 1 month at death |
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IV | None | 83 | 1 year 10 months at death |
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IIIc | None | 41 | 1 year 7 months ALC |
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IIIc | None | 52 | 1 year 9 months ALC |
| Chemonaïve | Carcinoma NOS | G3- poorly differentiated | IIc | None | 62 | 2 years 2 months ALC |
| Chemonaïve | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IIIc | None | 63 | 9 months ALC |
| Chemonaïve | Adenocarcinoma NOS | G3- poorly differentiated | IV | None | 52 | 1 year ALC |
| Recurrent | Serous cystadenocarcinoma NOS | G3- poorly differentiated | IV | Carboplatin and Paclitaxel 6 cycles Carboplatin 4 cycles Cisplatin 2 cycles Doxorubicin Pegylated Liposomal 1 cycle | 50 | 1 year 10 months at death |
| Recurrent | Serous surface papillary carcinoma (Gr3) | G3- poorly differentiated | IIIc | ICON6 Trial 8 cycles Carboplatin and Paclitaxel 6 cycles OVAR16/VEG110655 Trial 4 cycles Cisplatin 6 cycles Doxorubicin Pegylated Liposomal 6 cycles Docetaxel 3 cycles REZOLVE Study 3 cycles Paclitaxel 1 cycl | 59 | 4 years 7 months at death |
| Recurrent | Serous cystadenocarcinoma NOS (Gr3) | G3- poorly differentiated | IV | REZOLVE Study 5 cycles Unspecified Regimen 7 cycles Doxorubicin Pegylated Liposomal 6 cycles Paclitaxel 5 cycles Cisplatin 6 cycles | 57 | 5 years 8 months at death |
| Recurrent | Serous cystadenocarcinoma NOS (Gr3) | G3- poorly differentiated | IIIc | Carboplatin and Paclitaxel 6 cycles PrePARE Study 2 cycles Cisplatin 5 cycles Unspecified Regimen 6 cycles Doxorubicin Pegylated Liposomal 5 cycles | 61 | 2 years 2 months at death |
| Recurrent | Papillary serous adenocarcinoma (Gr2) | G1- well differentiated | IIIc | Carboplatin and Paclitaxel 3 cycles Doxorubicin Pegylated Liposomal 4 cycles | 31 | 9 months at death |
| Recurrent | Papillary serous adenocarcinoma (Gr2) | G3- poorly differentiated | IIc | Carboplatin and Paclitaxel 6 cycles Lilly Trial 3 cycles Cisplatin 1 cycle Doxorubicin Pegylated Liposomal 3 cycles Paclitaxel 2 cycles REZOLVE Study 1 cycle | 68 | 6 years 1 month at death |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadife, E.; Chan, E.; Luwor, R.; Kannourakis, G.; Findlay, J.; Ahmed, N. Paclitaxel-Induced Src Activation Is Inhibited by Dasatinib Treatment, Independently of Cancer Stem Cell Properties, in a Mouse Model of Ovarian Cancer. Cancers 2019, 11, 243. https://doi.org/10.3390/cancers11020243
Kadife E, Chan E, Luwor R, Kannourakis G, Findlay J, Ahmed N. Paclitaxel-Induced Src Activation Is Inhibited by Dasatinib Treatment, Independently of Cancer Stem Cell Properties, in a Mouse Model of Ovarian Cancer. Cancers. 2019; 11(2):243. https://doi.org/10.3390/cancers11020243
Chicago/Turabian StyleKadife, Elif, Emily Chan, Rodney Luwor, George Kannourakis, Jock Findlay, and Nuzhat Ahmed. 2019. "Paclitaxel-Induced Src Activation Is Inhibited by Dasatinib Treatment, Independently of Cancer Stem Cell Properties, in a Mouse Model of Ovarian Cancer" Cancers 11, no. 2: 243. https://doi.org/10.3390/cancers11020243
APA StyleKadife, E., Chan, E., Luwor, R., Kannourakis, G., Findlay, J., & Ahmed, N. (2019). Paclitaxel-Induced Src Activation Is Inhibited by Dasatinib Treatment, Independently of Cancer Stem Cell Properties, in a Mouse Model of Ovarian Cancer. Cancers, 11(2), 243. https://doi.org/10.3390/cancers11020243