Protein Phosphatases—A Touchy Enemy in the Battle Against Glioblastomas: A Review


Abstract
1. Introduction
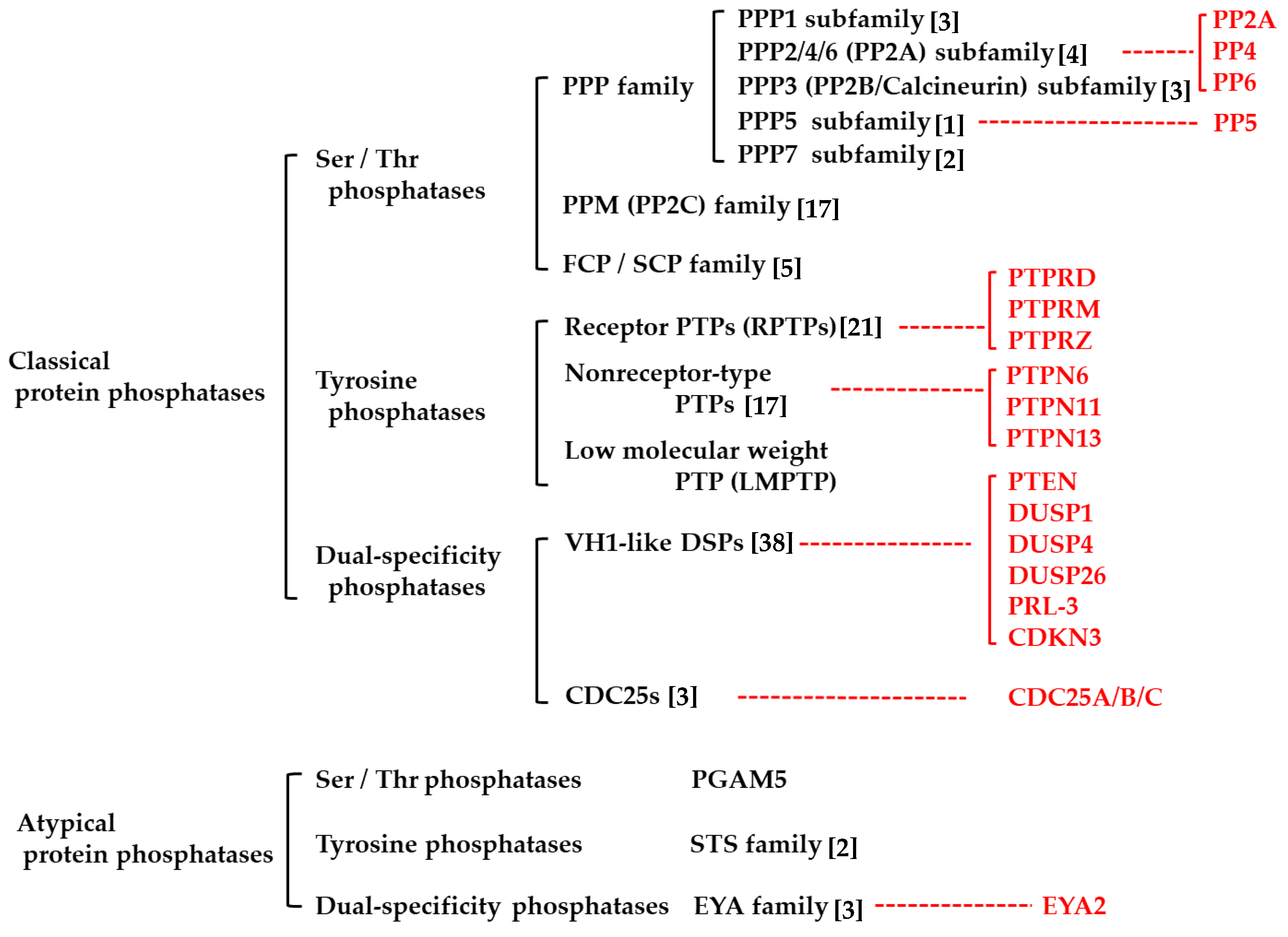
2. Protein Phosphatases
2.1. Classical Protein Phosphatases
2.1.1. Protein Serine/Threonine Phosphatases
Protein Phosphatase 2A
Protein Phosphatase 4
Protein Phosphatase 5
Protein Phosphatase 6
2.1.2. Protein Tyrosine Phosphatases
Tyrosine-Protein Phosphatase Non-Receptor Type 6
Tyrosine-Protein Phosphatase Non-Receptor Type 11
Tyrosine-Protein Phosphatase Non-Receptor Type 13
Receptor-Type Tyrosine-Protein Phosphatase Delta
Receptor-Type Tyrosine-Protein Phosphatase Mu
Receptor-Type Tyrosine-Protein Phosphatase Zeta
2.1.3. Dual-Specificity Phosphatases
Phosphatase and Tensin Homolog
Dual-Specificity Phosphatase 1/Mitogen-Activated Protein Kinase Phosphatase 1
Dual-Specificity Phosphatase 4/Mitogen-Activated Protein Kinase Phosphatase 2
Dual-Specificity Phosphatase 26/Mitogen-Activated Protein Kinase Phosphatase 8
Phosphatase of Regenerating Liver 3
Cyclin-Dependent Kinase Inhibitor 3
CDC25 Family
2.2. Atypical Protein Phosphatases
Eyes Absent Transcriptional Coactivator and Phosphatase Homolog 2
3. Phosphatase Targeting in GBM
3.1. PP2A Inhibitors and Activators
3.2. SHP2 Inhibitors and Activators
3.3. PTPRZ Inhibitor
3.4. DUSP1 Inhibitors
3.5. PRL-3 Inhibitors
3.6. CDC25s Inhibitors
3.7. EYA2 Inhibitor
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, P.; Aldape, K. World Health Organization 2016 Classification of Central Nervous System Tumors. Neurol. Clin. 2018, 36, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- deCarvalho, A.C.; Kim, H.; Poisson, L.M.; Winn, M.E.; Mueller, C.; Cherba, D.; Koeman, J.; Seth, S.; Protopopov, A.; Felicella, M.; et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat. Genet. 2018, 50, 708–717. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.M.; Kang, S.H.; Park, C.K.; Jung, S.; Park, E.S.; Lee, J.S.; Kim, S.H.; Woo, H.G. Recurrent Glioblastomas Reveal Molecular Subtypes Associated with Mechanistic Implications of Drug-Resistance. PLoS ONE 2015, 10, e0140528. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro-Oncology 2010, 12, 304–316. [Google Scholar] [CrossRef]
- Ma, C.; Zhao, G.; Cruz, M.H.; Siden, A.; Yakisich, J.S. Translational gap in glioma research. Anticancer Agents Med. Chem. 2014, 14, 1110–1120. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Venkatesan, S.; Lamfers, M.L.; Dirven, C.M.; Leenstra, S. Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol. 2016, 5, 77–90. [Google Scholar] [CrossRef]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef]
- Navis, A.C.; van den Eijnden, M.; Schepens, J.T.; Hooft van Huijsduijnen, R.; Wesseling, P.; Hendriks, W.J. Protein tyrosine phosphatases in glioma biology. Acta Neuropathol. 2010, 119, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Sadatomi, D.; Tanimura, S.; Ozaki, K.; Takeda, K. Atypical protein phosphatases: Emerging players in cellular signaling. Int. J. Mol. Sci. 2013, 14, 4596–4612. [Google Scholar] [CrossRef] [PubMed]
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef]
- Prabhakar, S.; Asuthkar, S.; Lee, W.; Chigurupati, S.; Zakharian, E.; Tsung, A.J.; Velpula, K.K. Targeting DUSPs in glioblastomas—Wielding a double-edged sword? Cell Biol. Int. 2014, 38, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef]
- Pulido, R.; Hooft van Huijsduijnen, R. Protein tyrosine phosphatases: Dual-specificity phosphatases in health and disease. FEBS J. 2008, 275, 848–866. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Thompson, J.J.; Williams, C.S. Protein Phosphatase 2A in the Regulation of Wnt Signaling, Stem Cells, and Cancer. Genes 2018, 9, 121. [Google Scholar] [CrossRef]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Grech, G.; Baldacchino, S.; Saliba, C.; Grixti, M.P.; Gauci, R.; Petroni, V.; Fenech, A.G.; Scerri, C. Deregulation of the protein phosphatase 2A, PP2A in cancer: Complexity and therapeutic options. Tumour Biol. 2016, 37, 11691–11700. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dube, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-mediated phosphorylation of the tyrosine phosphatase PRL-3 is required for PRL-3 promotion of Rho activation, motility and invasion. PLoS ONE 2013, 8, e64309. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.M.; Zimmerman, M.W.; Thompson, T.; Homanics, G.E.; Lazo, J.S.; Lagasse, E. Deletion of Ptp4a3 reduces clonogenicity and tumor-initiation ability of colitis-associated cancer cells in mice. Stem Cell Res. 2014, 13, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Foehr, E.D.; Lorente, G.; Vincent, V.; Nikolich, K.; Urfer, R. FAS associated phosphatase (FAP-1) blocks apoptosis of astrocytomas through dephosphorylation of FAS. J. Neurooncol. 2005, 74, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Antony, R.; Lukiw, W.J.; Bazan, N.G. Neuroprotectin D1 induces dephosphorylation of Bcl-xL in a PP2A-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J. Biol. Chem. 2010, 285, 18301–18308. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. The broken “off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef]
- Kauko, O.; O’Connor, C.M.; Kulesskiy, E.; Sangodkar, J.; Aakula, A.; Izadmehr, S.; Yetukuri, L.; Yadav, B.; Padzik, A.; Laajala, T.D.; et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med. 2018, 10, eaaq1093. [Google Scholar] [CrossRef] [PubMed]
- Allen-Petersen, B.L.; Risom, T.; Feng, Z.; Wang, Z.; Jenny, Z.P.; Thoma, M.C.; Pelz, K.R.; Morton, J.P.; Sansom, O.J.; Lopez, C.D.; et al. Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Denisova, O.V.; Qiao, X.; Jumppanen, M.; Peuhu, E.; Ahmed, S.U.; Raheem, O.; Haapasalo, H.; Eriksson, J.; Chalmers, A.J.; et al. PP2A Inhibitor PME-1 Drives Kinase Inhibitor Resistance in Glioma Cells. Cancer Res. 2016, 76, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Smith, A.M.; McDougall, F.; Carpenter, H.; Horan, M.; Neviani, P.; Powell, J.A.; Thomas, D.; Guthridge, M.A.; Perrotti, D.; et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res. 2010, 70, 5438–5447. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Dun, M.D.; Lee, E.M.; Harrison, C.; Kahl, R.; Flanagan, H.; Panicker, N.; Mashkani, B.; Don, A.S.; Morris, J.; et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget 2016, 7, 47465–47478. [Google Scholar] [CrossRef] [PubMed]
- Bockelman, C.; Lassus, H.; Hemmes, A.; Leminen, A.; Westermarck, J.; Haglund, C.; Butzow, R.; Ristimaki, A. Prognostic role of CIP2A expression in serous ovarian cancer. Br. J. Cancer 2011, 105, 989–995. [Google Scholar] [CrossRef]
- Colella, S.; Ohgaki, H.; Ruediger, R.; Yang, F.; Nakamura, M.; Fujisawa, H.; Kleihues, P.; Walter, G. Reduced expression of the Aalpha subunit of protein phosphatase 2A in human gliomas in the absence of mutations in the Aalpha and Abeta subunit genes. Int. J. Cancer 2001, 93, 798–804. [Google Scholar] [CrossRef]
- Fan, Y.L.; Chen, L.; Wang, J.; Yao, Q.; Wan, J.Q. Over expression of PPP2R2C inhibits human glioma cells growth through the suppression of mTOR pathway. FEBS Lett. 2013, 587, 3892–3897. [Google Scholar] [CrossRef]
- Pitre, A.; Davis, N.; Paul, M.; Orr, A.W.; Skalli, O. Synemin promotes AKT-dependent glioblastoma cell proliferation by antagonizing PP2A. Mol. Biol. Cell 2012, 23, 1243–1253. [Google Scholar] [CrossRef]
- Gursel, D.B.; Banu, M.A.; Berry, N.; Marongiu, R.; Burkhardt, J.K.; Kobylarz, K.; Kaplitt, M.G.; Rafii, S.; Boockvar, J.A. Tight regulation between cell survival and programmed cell death in GBM stem-like cells by EGFR/GSK3b/PP2A signaling. J. Neurooncol. 2015, 121, 19–29. [Google Scholar] [CrossRef]
- Palanichamy, K.; Kanji, S.; Gordon, N.; Thirumoorthy, K.; Jacob, J.R.; Litzenberg, K.T.; Patel, D.; Chakravarti, A. NNMT Silencing Activates Tumor Suppressor PP2A, Inactivates Oncogenic STKs, and Inhibits Tumor Forming Ability. Clin. Cancer Res. 2017, 23, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okkeri, J.; Pavic, K.; Wang, Z.; Kauko, O.; Halonen, T.; Sarek, G.; Ojala, P.M.; Rao, Z.; Xu, W.; et al. Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep. 2017, 18, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Westermarck, J. Regulation of protein phosphatase 2A (PP2A) tumor suppressor function by PME-1. Biochem. Soc. Trans. 2016, 44, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Chen, S.; Liu, F.; Li, B.; Khatoon, S.; Grundke-Iqbal, I.; Iqbal, K. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett. 2011, 585, 2653–2659. [Google Scholar] [CrossRef]
- Castigli, E.; Sciaccaluga, M.; Schiavoni, G.; Brozzi, F.; Fabiani, R.; Gorello, P.; Gianfranceschi, G.L. GL15 and U251 glioblastoma-derived human cell lines are peculiarly susceptible to induction of mitotic death by very low concentrations of okadaic acid. Oncol. Rep. 2006, 15, 463–470. [Google Scholar] [CrossRef]
- Lu, J.; Zhuang, Z.; Song, D.K.; Mehta, G.U.; Ikejiri, B.; Mushlin, H.; Park, D.M.; Lonser, R.R. The effect of a PP2A inhibitor on the nuclear receptor corepressor pathway in glioma. J. Neurosurg. 2010, 113, 225–233. [Google Scholar] [CrossRef]
- Li, J.Y.; Huang, J.Y.; Li, M.; Zhang, H.; Xing, B.; Chen, G.; Wei, D.; Gu, P.Y.; Hu, W.X. Anisomycin induces glioma cell death via down-regulation of PP2A catalytic subunit in vitro. Acta Pharmacol. Sin. 2012, 33, 935–940. [Google Scholar] [CrossRef]
- Lu, J.; Kovach, J.S.; Johnson, F.; Chiang, J.; Hodes, R.; Lonser, R.; Zhuang, Z. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 11697–11702. [Google Scholar] [CrossRef]
- Gordon, I.K.; Lu, J.; Graves, C.A.; Huntoon, K.; Frerich, J.M.; Hanson, R.H.; Wang, X.; Hong, C.S.; Ho, W.; Feldman, M.J.; et al. Protein Phosphatase 2A Inhibition with LB100 Enhances Radiation-Induced Mitotic Catastrophe and Tumor Growth Delay in Glioblastoma. Mol. Cancer Ther. 2015, 14, 1540–1547. [Google Scholar] [CrossRef]
- Lankoff, A.; Bialczyk, J.; Dziga, D.; Carmichael, W.W.; Gradzka, I.; Lisowska, H.; Kuszewski, T.; Gozdz, S.; Piorun, I.; Wojcik, A. The repair of gamma-radiation-induced DNA damage is inhibited by microcystin-LR, the PP1 and PP2A phosphatase inhibitor. Mutagenesis 2006, 21, 83–90. [Google Scholar] [CrossRef]
- Lin, S.S.; Bassik, M.C.; Suh, H.; Nishino, M.; Arroyo, J.D.; Hahn, W.C.; Korsmeyer, S.J.; Roberts, T.M. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. J. Biol. Chem. 2006, 281, 23003–23012. [Google Scholar] [CrossRef] [PubMed]
- Scheidtmann, K.H.; Mumby, M.C.; Rundell, K.; Walter, G. Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: Inhibition by small-t antigen. Mol. Cell. Biol. 1991, 11, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Cai, X.; Shouse, G.P.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007, 26, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, Y.; Sheng, K.; Fei, X.; Guo, Q.; Larner, J.; Kong, X.; Qiu, Y.; Mi, J. Serine/threonine protein phosphatase 6 modulates the radiation sensitivity of glioblastoma. Cell Death Dis. 2011, 2, e241. [Google Scholar] [CrossRef] [PubMed]
- Ohama, T. The multiple functions of protein phosphatase 6. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T.; Philp, A.; Vazquez-Martin, C. Protein phosphatase 4—From obscurity to vital functions. FEBS Lett. 2005, 579, 3278–3286. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Xu, S.; Xue, P.; Li, Q.; Lu, Q.; Jia, Q.; Zhang, L.; Li, X.; Li, X. Protein phosphatase 4 catalytic subunit is overexpressed in glioma and promotes glioma cell proliferation and invasion. Tumour Biol. 2016, 37, 11893–11901. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, L.; Huang, L.; Ma, X.; Li, D.; Cai, S. High expression of protein phosphatase 4 is associated with the aggressive malignant behavior of colorectal carcinoma. Mol. Cancer 2015, 14, 95. [Google Scholar] [CrossRef]
- Weng, S.; Wang, H.; Chen, W.; Katz, M.H.; Chatterjee, D.; Lee, J.E.; Pisters, P.W.; Gomez, H.F.; Abbruzzese, J.L.; Fleming, J.B.; et al. Overexpression of protein phosphatase 4 correlates with poor prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1336–1343. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, A.; Sun, L.; Zhong, X.; Zhong, J.; Wang, H.; Cai, M.; Li, J.; Xu, Y.; Liao, J.; et al. Protein phosphatase PP4 is overexpressed in human breast and lung tumors. Cell Res. 2008, 18, 974–977. [Google Scholar] [CrossRef]
- Becker, W.; Kentrup, H.; Klumpp, S.; Schultz, J.E.; Joost, H.G. Molecular cloning of a protein serine/threonine phosphatase containing a putative regulatory tetratricopeptide repeat domain. J. Biol. Chem. 1994, 269, 22586–22592. [Google Scholar] [PubMed]
- Chinkers, M. Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 11075–11079. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Sanchez, E.R. Protein phosphatase 5. Int. J. Biochem. Cell Biol. 2008, 40, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Atiye, J.; Wolf, M.; Kaur, S.; Monni, O.; Bohling, T.; Kivioja, A.; Tas, E.; Serra, M.; Tarkkanen, M.; Knuutila, S. Gene amplifications in osteosarcoma-CGH microarray analysis. Genes Chromosomes Cancer 2005, 42, 158–163. [Google Scholar] [CrossRef]
- Golden, T.; Swingle, M.; Honkanen, R.E. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev. 2008, 27, 169–178. [Google Scholar] [CrossRef]
- Zhi, X.; Zhang, H.; He, C.; Wei, Y.; Bian, L.; Li, G. Serine/Threonine Protein Phosphatase-5 Accelerates Cell Growth and Migration in Human Glioma. Cell. Mol. Neurobiol. 2015, 35, 669–677. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Schlosser, K.A.; Lyons, S.P.; Nasa, I.; Gui, J.; Adamo, M.E.; Gerber, S.A. Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal. 2018, 11, eaaq1441. [Google Scholar] [CrossRef]
- Rusin, S.F.; Adamo, M.E.; Kettenbach, A.N. Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2017, 5, 97. [Google Scholar] [CrossRef]
- Sooman, L.; Ekman, S.; Tsakonas, G.; Jaiswal, A.; Navani, S.; Edqvist, P.H.; Ponten, F.; Bergstrom, S.; Johansson, M.; Wu, X.; et al. PTPN6 expression is epigenetically regulated and influences survival and response to chemotherapy in high-grade gliomas. Tumour Biol. 2014, 35, 4479–4488. [Google Scholar] [CrossRef]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Matsumoto, K.; Nishida, E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J. Biol. Chem. 2004, 279, 22992–22995. [Google Scholar] [CrossRef]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, M.L. Key insights into the protein tyrosine phosphatase PTPN11/SHP2 associated with noonan syndrome and cancer. Hum. Mutat. 2017, 38, 337. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, L.A.; Toering, S.J.; Simon, M.A.; Krasnow, M.A.; Smith-Bolton, R.K. Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development 2006, 133, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed]
- Lauriol, J.; Jaffre, F.; Kontaridis, M.I. The role of the protein tyrosine phosphatase SHP2 in cardiac development and disease. Semin. Cell Dev. Biol. 2015, 37, 73–81. [Google Scholar] [CrossRef]
- Zheng, H.; Yu, W.M.; Waclaw, R.R.; Kontaridis, M.I.; Neel, B.G.; Qu, C.K. Gain-of-function mutations in the gene encoding the tyrosine phosphatase SHP2 induce hydrocephalus in a catalytically dependent manner. Sci. Signal. 2018, 11, eaao1591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chan, R.J.; Chen, H.; Yang, Z.; He, Y.; Zhang, X.; Luo, Y.; Yin, F.; Moh, A.; Miller, L.C.; et al. Negative regulation of Stat3 by activating PTPN11 mutants contributes to the pathogenesis of Noonan syndrome and juvenile myelomonocytic leukemia. J. Biol. Chem. 2009, 284, 22353–22363. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Li, S.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef]
- Zhan, Y.; Counelis, G.J.; O’Rourke, D.M. The protein tyrosine phosphatase SHP-2 is required for EGFRvIII oncogenic transformation in human glioblastoma cells. Exp. Cell Res. 2009, 315, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Mu, L.; Qiao, W.; Li, H.; Tang, J.; Wang, C.; Hu, W.; Zhao, T.; Dong, B.; Song, Y.; et al. Inhibition of SHP-2 promotes radiosensitivity in glioma. Mol. Med. Rep. 2015, 12, 3563–3568. [Google Scholar] [CrossRef] [PubMed]
- Saras, J.; Franzen, P.; Aspenstrom, P.; Hellman, U.; Gonez, L.J.; Heldin, C.H. A novel GTPase-activating protein for Rho interacts with a PDZ domain of the protein-tyrosine phosphatase PTPL1. J. Biol. Chem. 1997, 272, 24333–24338. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; Anderson, M.E.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.J.; Lee, J.H. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Abaan, O.D.; Levenson, A.; Khan, O.; Furth, P.A.; Uren, A.; Toretsky, J.A. PTPL1 is a direct transcriptional target of EWS-FLI1 and modulates Ewing’s Sarcoma tumorigenesis. Oncogene 2005, 24, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Revillion, F.; Puech, C.; Rabenoelina, F.; Chalbos, D.; Peyrat, J.P.; Freiss, G. Expression of the putative tumor suppressor gene PTPN13/PTPL1 is an independent prognostic marker for overall survival in breast cancer. Int. J. Cancer 2009, 124, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Abaan, O.D.; Toretsky, J.A. PTPL1: A large phosphatase with a split personality. Cancer Metastasis Rev. 2008, 27, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bei, L.; Eklund, E.A. Inhibition of Fas associated phosphatase 1 (Fap1) facilitates apoptosis of colon cancer stem cells and enhances the effects of oxaliplatin. Oncotarget 2018, 9, 25891–25902. [Google Scholar] [CrossRef]
- Shinoura, N.; Yamamoto, N.; Asai, A.; Kirino, T.; Hamada, H. Adenovirus-mediated transfer of Fas ligand gene augments radiation-induced apoptosis in U-373MG glioma cells. Jpn. J. Cancer Res. 2000, 91, 1044–1050. [Google Scholar] [CrossRef]
- Maleniak, T.C.; Darling, J.L.; Lowenstein, P.R.; Castro, M.G. Adenovirus-mediated expression of HSV1-TK or Fas ligand induces cell death in primary human glioma-derived cell cultures that are resistant to the chemotherapeutic agent CCNU. Cancer Gene Ther. 2001, 8, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Brito, M.R.; Bixby, J.L. Differential activities in adhesion and neurite growth of fibronectin type III repeats in the PTP-delta extracellular domain. Int. J. Dev. Neurosci. 2006, 24, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Choucair, N.; Mignon-Ravix, C.; Cacciagli, P.; Abou Ghoch, J.; Fawaz, A.; Megarbane, A.; Villard, L.; Chouery, E. Evidence that homozygous PTPRD gene microdeletion causes trigonocephaly, hearing loss, and intellectual disability. Mol. Cytogenet. 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Uetani, N.; Kato, K.; Ogura, H.; Mizuno, K.; Kawano, K.; Mikoshiba, K.; Yakura, H.; Asano, M.; Iwakura, Y. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000, 19, 2775–2785. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef]
- Solomon, D.A.; Kim, J.S.; Cronin, J.C.; Sibenaller, Z.; Ryken, T.; Rosenberg, S.A.; Ressom, H.; Jean, W.; Bigner, D.; Yan, H.; et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008, 68, 10300–10306. [Google Scholar] [CrossRef]
- Ortiz, B.; Fabius, A.W.; Wu, W.H.; Pedraza, A.; Brennan, C.W.; Schultz, N.; Pitter, K.L.; Bromberg, J.F.; Huse, J.T.; Holland, E.C.; et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 8149–8154. [Google Scholar] [CrossRef]
- Brady-Kalnay, S.M.; Tonks, N.K. Identification of the homophilic binding site of the receptor protein tyrosine phosphatase PTP mu. J. Biol. Chem. 1994, 269, 28472–28477. [Google Scholar]
- Sudhir, P.R.; Lin, S.T.; Chia-Wen, C.; Yang, S.H.; Li, A.F.; Lai, R.H.; Wang, M.J.; Chen, Y.T.; Chen, C.F.; Jou, Y.S.; et al. Loss of PTPRM associates with the pathogenic development of colorectal adenoma-carcinoma sequence. Sci. Rep. 2015, 5, 9633. [Google Scholar] [CrossRef]
- Sun, P.H.; Ye, L.; Mason, M.D.; Jiang, W.G. Protein tyrosine phosphatase micro (PTP micro or PTPRM), a negative regulator of proliferation and invasion of breast cancer cells, is associated with disease prognosis. PLoS ONE 2012, 7, e50183. [Google Scholar] [CrossRef]
- Burgoyne, A.M.; Palomo, J.M.; Phillips-Mason, P.J.; Burden-Gulley, S.M.; Major, D.L.; Zaremba, A.; Robinson, S.; Sloan, A.E.; Vogelbaum, M.A.; Miller, R.H.; et al. PTPmu suppresses glioma cell migration and dispersal. Neuro-Oncology 2009, 11, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.M.; Phillips-Mason, P.J.; Burden-Gulley, S.M.; Robinson, S.; Sloan, A.E.; Miller, R.H.; Brady-Kalnay, S.M. Proteolytic cleavage of protein tyrosine phosphatase mu regulates glioblastoma cell migration. Cancer Res. 2009, 69, 6960–6968. [Google Scholar] [CrossRef] [PubMed]
- Krueger, N.X.; Saito, H. A human transmembrane protein-tyrosine-phosphatase, PTP zeta, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc. Natl. Acad. Sci. USA 1992, 89, 7417–7421. [Google Scholar] [CrossRef]
- Ariyama, T.; Hasegawa, K.; Inazawa, J.; Mizuno, K.; Ogimoto, M.; Katagiri, T.; Yakura, H. Assignment of the human protein tyrosine phosphatase, receptor-type, zeta (PTPRZ) gene to chromosome band 7q31.3. Cytogenet. Cell Genet. 1995, 70, 52–54. [Google Scholar] [CrossRef]
- Canoll, P.D.; Petanceska, S.; Schlessinger, J.; Musacchio, J.M. Three forms of RPTP-beta are differentially expressed during gliogenesis in the developing rat brain and during glial cell differentiation in culture. J. Neurosci. Res. 1996, 44, 199–215. [Google Scholar] [CrossRef]
- Maurel, P.; Rauch, U.; Flad, M.; Margolis, R.K.; Margolis, R.U. Phosphacan, a chondroitin sulfate proteoglycan of brain that interacts with neurons and neural cell-adhesion molecules, is an extracellular variant of a receptor-type protein tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1994, 91, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kunkel, P.; Lamszus, K.; Ulbricht, U.; Lorente, G.A.; Nelson, A.M.; von Schack, D.; Chin, D.J.; Lohr, S.C.; Westphal, M.; et al. A role for receptor tyrosine phosphatase zeta in glioma cell migration. Oncogene 2003, 22, 6661–6668. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, U.; Brockmann, M.A.; Aigner, A.; Eckerich, C.; Muller, S.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Expression and function of the receptor protein tyrosine phosphatase zeta and its ligand pleiotrophin in human astrocytomas. J. Neuropathol. Exp. Neurol. 2003, 62, 1265–1275. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.M.; Navis, A.C.; Schepens, J.T.; Verrijp, K.; Hovestad, L.; Hilhorst, R.; Harroch, S.; Wesseling, P.; Leenders, W.P.; Hendriks, W.J. Intracellular and extracellular domains of protein tyrosine phosphatase PTPRZ-B differentially regulate glioma cell growth and motility. Oncotarget 2014, 5, 8690–8702. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, U.; Eckerich, C.; Fillbrandt, R.; Westphal, M.; Lamszus, K. RNA interference targeting protein tyrosine phosphatase zeta/receptor-type protein tyrosine phosphatase beta suppresses glioblastoma growth in vitro and in vivo. J. Neurochem. 2006, 98, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Tohma, Y.; Gratas, C.; Biernat, W.; Peraud, A.; Fukuda, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Phillips, J.; Onar-Thomas, A.; Romero, E.; Zheng, S.; Wiencke, J.K.; McBride, S.M.; Cowdrey, C.; Prados, M.D.; Weiss, W.A.; et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro-Oncology 2012, 14, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef]
- Godlewski, J.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A. Belonging to a network—MicroRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro-Oncology 2015, 17, 652–662. [Google Scholar] [CrossRef]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Li, M.; Zhou, J.Y.; Ge, Y.; Matherly, L.H.; Wu, G.S. The phosphatase MKP1 is a transcriptional target of p53 involved in cell cycle regulation. J. Biol. Chem. 2003, 278, 41059–41068. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Park, J.; Lee, J.; Choi, K.; Choi, C. Constitutive Expression of MAP Kinase Phosphatase-1 Confers Multi-drug Resistance in Human Glioblastoma Cells. Cancer Res. Treat. 2012, 44, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.L.; Kahana, S.; Blass, M.; Brody, Y.; Okhrimenko, H.; Xiang, C.; Finniss, S.; Blumberg, P.M.; Lee, H.K.; Brodie, C. Phosphorylation of protein kinase Cdelta on distinct tyrosine residues induces sustained activation of Erk1/2 via down-regulation of MKP-1: Role in the apoptotic effect of etoposide. J. Biol. Chem. 2008, 283, 17731–17739. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Lin, A.; Marshall, M.S. Pancreatic tumor cells with mutant K-ras suppress ERK activity by MEK-dependent induction of MAP kinase phosphatase-2. Biochem. Biophys. Res. Commun. 2001, 280, 992–997. [Google Scholar] [CrossRef]
- Wang, H.Y.; Cheng, Z.; Malbon, C.C. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003, 191, 229–237. [Google Scholar] [CrossRef]
- Gaedcke, J.; Grade, M.; Jung, K.; Camps, J.; Jo, P.; Emons, G.; Gehoff, A.; Sax, U.; Schirmer, M.; Becker, H.; et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer 2010, 49, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Sieben, N.L.; Oosting, J.; Flanagan, A.M.; Prat, J.; Roemen, G.M.; Kolkman-Uljee, S.M.; van Eijk, R.; Cornelisse, C.J.; Fleuren, G.J.; van Engeland, M. Differential gene expression in ovarian tumors reveals Dusp 4 and Serpina 5 as key regulators for benign behavior of serous borderline tumors. J. Clin. Oncol. 2005, 23, 7257–7264. [Google Scholar] [CrossRef] [PubMed]
- Chitale, D.; Gong, Y.; Taylor, B.S.; Broderick, S.; Brennan, C.; Somwar, R.; Golas, B.; Wang, L.; Motoi, N.; Szoke, J.; et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 2009, 28, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Waha, A.; Felsberg, J.; Hartmann, W.; von dem Knesebeck, A.; Mikeska, T.; Joos, S.; Wolter, M.; Koch, A.; Yan, P.S.; Endl, E.; et al. Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010, 70, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Gessi, M.; Waha, A.; Isselstein, L.J.; Luxen, D.; Freihoff, D.; Freihoff, J.; Becker, A.; Simon, M.; Hammes, J.; et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am. J. Pathol. 2012, 181, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-Oncology 2018, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Hoshide, R.; Jandial, R. 2016 World Health Organization Classification of Central Nervous System Tumors: An Era of Molecular Biology. World Neurosurg. 2016, 94, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Vasudevan, S.A.; Yu, Y.; Ge, N.; Ludwig, A.D.; Wesson, C.L.; Wang, K.; Burlingame, S.M.; Zhao, Y.J.; Rao, P.H.; et al. Dual-specificity phosphatase 26 is a novel p53 phosphatase and inhibits p53 tumor suppressor functions in human neuroblastoma. Oncogene 2010, 29, 4938–4946. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.M.; Verrijp, K.; Schepens, J.T.; Navis, A.C.; Piepers, J.A.; Palmen, C.B.; van den Eijnden, M.; Hooft van Huijsduijnen, R.; Wesseling, P.; Leenders, W.P.; et al. Comprehensive protein tyrosine phosphatase mRNA profiling identifies new regulators in the progression of glioma. Acta Neuropathol. Commun. 2016, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, N.; Nomura, M.; Ikeda, M.; Kasugai, I.; Tsubaki, Y.; Takagaki, K.; Kawamura, T.; Yamashita, Y.; Sato, I.; Sato, M.; et al. Protein phosphatase Dusp26 associates with KIF3 motor and promotes N-cadherin-mediated cell-cell adhesion. Oncogene 2009, 28, 752–761. [Google Scholar] [CrossRef]
- Wang, H.; Quah, S.Y.; Dong, J.M.; Manser, E.; Tang, J.P.; Zeng, Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007, 67, 2922–2926. [Google Scholar] [CrossRef]
- Abdollahi, P.; Vandsemb, E.N.; Hjort, M.A.; Misund, K.; Holien, T.; Sponaas, A.M.; Ro, T.B.; Slordahl, T.S.; Borset, M. Src Family Kinases Are Regulated in Multiple Myeloma Cells by Phosphatase of Regenerating Liver-3. Mol. Cancer Res. 2017, 15, 69–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Fan, X.; Xiong, J.; Zhang, G.; Luo, X.; Li, K.; Jie, Z.; Cao, Y.; Huang, Z.; et al. PRL-3 promotes gastric cancer peritoneal metastasis via the PI3K/AKT signaling pathway in vitro and in vivo. Oncol. Lett. 2018, 15, 9069–9074. [Google Scholar] [CrossRef] [PubMed]
- Li, B.H.; Wang, Y.; Wang, C.Y.; Zhao, M.J.; Deng, T.; Ren, X.Q. Up-Regulation of Phosphatase in Regenerating Liver-3 (PRL-3) Contributes to Malignant Progression of Hepatocellular Carcinoma by Activating Phosphatase and Tensin Homolog Deleted on Chromosome Ten (PTEN)/Phosphoinositide 3-Kinase (PI3K)/AKT Signaling Pathway. Med. Sci. Monit. 2018, 24, 8105–8114. [Google Scholar] [PubMed]
- Xie, H.; Wang, H. PRL-3 promotes breast cancer progression by downregulating p14(ARF)-mediated p53 expression. Oncol. Lett. 2018, 15, 2795–2800. [Google Scholar] [PubMed]
- Vandsemb, E.N.; Bertilsson, H.; Abdollahi, P.; Storkersen, O.; Vatsveen, T.K.; Rye, M.B.; Ro, T.B.; Borset, M.; Slordahl, T.S. Phosphatase of regenerating liver 3 (PRL-3) is overexpressed in human prostate cancer tissue and promotes growth and migration. J. Transl. Med. 2016, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Mu, N.; Gu, J.; Liu, N.; Xue, X.; Shu, Z.; Zhang, K.; Huang, T.; Chu, C.; Zhang, W.; Gong, L.; et al. PRL-3 is a potential glioblastoma prognostic marker and promotes glioblastoma progression by enhancing MMP7 through the ERK and JNK pathways. Theranostics 2018, 8, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Soni, P.; Husain, N.; Chandra, A.; Ojha, B.K.; Bhatt, M.L.; Gupta, R.K. Do phosphatase of regenerating liver-3, matrix metalloproteinases-2, matrix metalloproteinases-9, and epidermal growth factor receptor-1 predict response to therapy and survival in glioblastoma multiforme? Indian J. Pathol. Microbiol. 2016, 59, 287–293. [Google Scholar] [PubMed]
- Kong, L.; Li, Q.; Wang, L.; Liu, Z.; Sun, T. The value and correlation between PRL-3 expression and matrix metalloproteinase activity and expression in human gliomas. Neuropathology 2007, 27, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Gyuris, J.; Golemis, E.; Chertkov, H.; Brent, R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 1993, 75, 791–803. [Google Scholar] [CrossRef]
- Hannon, G.J.; Casso, D.; Beach, D. KAP: A dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 1731–1735. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, X.; Schoch, B.S.; Carroll, R.S.; Black, P.M.; Johnson, M.D. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007, 67, 130–138. [Google Scholar] [CrossRef]
- Li, H.; Jiang, X.; Yu, Y.; Huang, W.; Xing, H.; Agar, N.Y.; Yang, H.W.; Yang, B.; Carroll, R.S.; Johnson, M.D. KAP regulates ROCK2 and Cdk2 in an RNA-activated glioblastoma invasion pathway. Oncogene 2015, 34, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Kasugai, I.; Sato, M.; Tanuma, N.; Sato, I.; Nomura, M.; Yamashita, K.; Sonoda, Y.; Kumabe, T.; Tominaga, T.; et al. CDC25A mRNA levels significantly correlate with Ki-67 expression in human glioma samples. J. Neurooncol. 2010, 100, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Zhang, Y.; Xia, Y.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7, 12431. [Google Scholar] [CrossRef] [PubMed]
- Vanan, I.; Dong, Z.; Tosti, E.; Warshaw, G.; Symons, M.; Ruggieri, R. Role of a DNA damage checkpoint pathway in ionizing radiation-induced glioblastoma cell migration and invasion. Cell. Mol. Neurobiol. 2012, 32, 1199–1208. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, S.; Zhen, Y.; Li, Q.; Teng, L.; Asai, A.; Kawamoto, K. A miR-21 inhibitor enhances apoptosis and reduces G(2)-M accumulation induced by ionizing radiation in human glioblastoma U251 cells. Brain Tumor Pathol. 2011, 28, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Suren, D.; Isiksacan Ozen, O. CDC25B, Ki-67, and p53 expressions in reactive gliosis and astrocytomas. J. BUON 2013, 18, 1006–1011. [Google Scholar]
- Nakabayashi, H.; Hara, M.; Shimizu, K. Prognostic significance of CDC25B expression in gliomas. J. Clin. Pathol. 2006, 59, 725–728. [Google Scholar] [CrossRef]
- Niu, M.; Cai, W.; Liu, H.; Chong, Y.; Hu, W.; Gao, S.; Shi, Q.; Zhou, X.; Liu, X.; Yu, R. Plumbagin inhibits growth of gliomas in vivo via suppression of FOXM1 expression. J. Pharmacol. Sci. 2015, 128, 131–136. [Google Scholar] [CrossRef]
- Liu, X.; Cai, W.; Niu, M.; Chong, Y.; Liu, H.; Hu, W.; Wang, D.; Gao, S.; Shi, Q.; Hu, J.; et al. Plumbagin induces growth inhibition of human glioma cells by downregulating the expression and activity of FOXM1. J. Neurooncol. 2015, 121, 469–477. [Google Scholar] [CrossRef]
- Jung, Y.; Joo, K.M.; Seong, D.H.; Choi, Y.L.; Kong, D.S.; Kim, Y.; Kim, M.H.; Jin, J.; Suh, Y.L.; Seol, H.J.; et al. Identification of prognostic biomarkers for glioblastomas using protein expression profiling. Int. J. Oncol. 2012, 40, 1122–1132. [Google Scholar] [CrossRef]
- Lal, N.; Nemaysh, V.; Luthra, P.M. Proteasome mediated degradation of CDC25C and Cyclin B1 in Demethoxycurcumin treated human glioma U87 MG cells to trigger G2/M cell cycle arrest. Toxicol. Appl. Pharmacol. 2018, 356, 76–89. [Google Scholar] [CrossRef]
- Garcia-Morales, P.; Carrasco-Garcia, E.; Ruiz-Rico, P.; Martinez-Mira, R.; Menendez-Gutierrez, M.P.; Ferragut, J.A.; Saceda, M.; Martinez-Lacaci, I. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene 2007, 26, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Silver, S.J.; Davies, E.L.; Doyon, L.; Rebay, I. Functional dissection of eyes absent reveals new modes of regulation within the retinal determination gene network. Mol. Cell. Biol. 2003, 23, 5989–5999. [Google Scholar] [CrossRef]
- Kumar, J.P.; Moses, K. EGF receptor and Notch signaling act upstream of Eyeless/Pax6 to control eye specification. Cell 2001, 104, 687–697. [Google Scholar] [CrossRef]
- Kenyon, K.L.; Ranade, S.S.; Curtiss, J.; Mlodzik, M.; Pignoni, F. Coordinating proliferation and tissue specification to promote regional identity in the Drosophila head. Dev. Cell 2003, 5, 403–414. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Jeong, D.G.; Jung, S.K.; Ryu, S.E.; Xiao, A.; Allis, C.D.; Kim, S.J.; Tonks, N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J. Biol. Chem. 2009, 284, 16066–16070. [Google Scholar] [CrossRef]
- Farabaugh, S.M.; Micalizzi, D.S.; Jedlicka, P.; Zhao, R.; Ford, H.L. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 2012, 31, 552–562. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, N.; Huang, J.; Buckanovich, R.J.; Liang, S.; Barchetti, A.; Vezzani, C.; O’Brien-Jenkins, A.; Wang, J.; Ward, M.R.; et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005, 65, 925–932. [Google Scholar] [PubMed]
- Wen, Z.; Liang, C.; Pan, Q.; Wang, Y. Eya2 overexpression promotes the invasion of human astrocytoma through the regulation of ERK/MMP9 signaling. Int. J. Mol. Med. 2017, 40, 1315–1322. [Google Scholar] [CrossRef]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Yamamoto, D.; Sakurai, S.; Hasegawa, M.; Aizu-Yokota, E.; Momoi, T.; Kasahara, T. FTY720, a novel immunosuppressive agent, induces apoptosis in human glioma cells. Biochem. Biophys. Res. Commun. 2001, 281, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Kastrinsky, D.B.; Sangodkar, J.; Zaware, N.; Izadmehr, S.; Dhawan, N.S.; Narla, G.; Ohlmeyer, M. Reengineered tricyclic anti-cancer agents. Bioorg. Med. Chem. 2015, 23, 6528–6534. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Perl, A.; Tohme, R.; Kiselar, J.; Kastrinsky, D.B.; Zaware, N.; Izadmehr, S.; Mazhar, S.; Wiredja, D.D.; O’Connor, C.M.; et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Investig. 2017, 127, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yu, B.; Xu, G.; Xu, W.R.; Loh, M.L.; Tang, L.D.; Qu, C.K. Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J. Med. Chem. 2013, 56, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, S.; Li, C.; Zhou, C.; Li, D.; Liu, P.; Huang, M.; Shen, X. Cryptotanshinone inhibits human glioma cell proliferation in vitro and in vivo through SHP-2-dependent inhibition of STAT3 activation. Cell Death Dis. 2017, 8, e2767. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, C.; Li, D.; Wang, Y.; Zhou, C.; Shao, W.; Peng, J.; You, Y.; Zhang, X.; Shen, X. Cryptotanshinone inhibits human glioma cell proliferation by suppressing STAT3 signaling. Mol. Cell. Biochem. 2013, 381, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, R.; Ran, H.; Neel, B.G. Off-target inhibition by active site-targeting SHP2 inhibitors. FEBS Open Bio 2018, 8, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lu, Y.; Chen, G.; Huang, S. Molecular evidence of cryptotanshinone for treatment and prevention of human cancer. Anticancer Agents Med. Chem. 2013, 13, 979–987. [Google Scholar] [CrossRef]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res.. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Uciechowski, P.; Fenchel, K.; Chevassut, T. Targeting SHP-1, 2 and SHIP Pathways: A Novel Strategy for Cancer Treatment? Oncology 2018, 95, 257–269. [Google Scholar] [CrossRef]
- Fujikawa, A.; Nagahira, A.; Sugawara, H.; Ishii, K.; Imajo, S.; Matsumoto, M.; Kuboyama, K.; Suzuki, R.; Tanga, N.; Noda, M.; et al. Small-molecule inhibition of PTPRZ reduces tumor growth in a rat model of glioblastoma. Sci. Rep. 2016, 6, 20473. [Google Scholar] [CrossRef]
- Fujikawa, A.; Sugawara, H.; Tanaka, T.; Matsumoto, M.; Kuboyama, K.; Suzuki, R.; Tanga, N.; Ogata, A.; Masumura, M.; Noda, M. Targeting PTPRZ inhibits stem cell-like properties and tumorigenicity in glioblastoma cells. Sci. Rep. 2017, 7, 5609. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, W.; Su, X.; Wu, S.; Lin, Y.; Li, J.; Wang, Y.; Chen, J.; Zhou, Y.; Qiu, P.; et al. Triptolide inhibits proliferation and invasion of malignant glioma cells. J. Neurooncol. 2012, 109, 53–62. [Google Scholar] [CrossRef]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; Queiroz de Oliveira, P.; Znosko, W.; Smithgall, T.E.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Kaltenmeier, C.T.; Vollmer, L.L.; Vernetti, L.A.; Caprio, L.; Davis, K.; Korotchenko, V.N.; Day, B.W.; Tsang, M.; Hulkower, K.I.; Lotze, M.T.; et al. A Tumor Cell-Selective Inhibitor of Mitogen-Activated Protein Kinase Phosphatases Sensitizes Breast Cancer Cells to Lymphokine-Activated Killer Cell Activity. J. Pharmacol. Exp. Ther. 2017, 361, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.N.; Liao, Y.F.; Lu, Y.X.; Wang, Y.; Lu, J.H.; Zeng, Z.L.; Huang, Q.T.; Sheng, H.; Yun, J.P.; Xie, D.; et al. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 2018, 412, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Daouti, S.; Li, W.H.; Qian, H.; Huang, K.S.; Holmgren, J.; Levin, W.; Reik, L.; McGady, D.L.; Gillespie, P.; Perrotta, A.; et al. A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130Cas cleavage. Cancer Res. 2008, 68, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yu, Z.H.; Liu, S.; Zhang, L.; Zhang, R.Y.; Zeng, L.F.; Zhang, S.; Zhang, Z.Y. Novel Anticancer Agents Based on Targeting the Trimer Interface of the PRL Phosphatase. Cancer Res. 2016, 76, 4805–4815. [Google Scholar] [CrossRef] [PubMed]
- Thura, M.; Al-Aidaroos, A.Q.; Yong, W.P.; Kono, K.; Gupta, A.; Lin, Y.B.; Mimura, K.; Thiery, J.P.; Goh, B.C.; Tan, P.; et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight 2016, 1, e87607. [Google Scholar] [CrossRef] [PubMed]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinkovic, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, C.P.; Burkhardt, J.K.; Shin, B.J.; Gursel, D.B.; Mubita, L.; Gorrepati, R.; Brennan, C.; Holland, E.C.; Boockvar, J.A. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PLoS ONE 2012, 7, e30059. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Pan, L.; Groen, R.W.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Investig. 2014, 124, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Li, J.R.; Wu, C.C.; Wang, J.D.; Yang, C.P.; Chen, W.Y.; Wang, W.Y.; Chen, C.J. Indomethacin induced glioma apoptosis involving ceramide signals. Exp. Cell Res. 2018, 365, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sung, S.S.; Yip, M.L.; Lawrence, H.R.; Ren, Y.; Guida, W.C.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 2006, 70, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, K.; Grosskopf, S.; Lum, C.T.; Wurtele, M.; Roder, N.; von Kries, J.P.; Rosario, M.; Rademann, J.; Birchmeier, W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. USA 2008, 105, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yoon, Y.J.; Jeon, Y.J.; Choi, J.; Lee, Y.J.; Lee, J.; Choi, S.; Nash, O.; Han, D.C.; Kwon, B.M. Geranylnaringenin (CG902) inhibits constitutive and inducible STAT3 activation through the activation of SHP-2 tyrosine phosphatase. Biochem. Pharmacol. 2017, 142, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, A.; Ichimura, K. Signal transduction pathways and resistance to targeted therapies in glioma. Semin. Cancer Biol. 2019. [Google Scholar]
- Krueger, A.B.; Drasin, D.J.; Lea, W.A.; Patrick, A.N.; Patnaik, S.; Backos, D.S.; Matheson, C.J.; Hu, X.; Barnaeva, E.; Holliday, M.J.; et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J. Biol. Chem. 2014, 289, 16349–16361. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomiyama, A.; Kobayashi, T.; Mori, K.; Ichimura, K. Protein Phosphatases—A Touchy Enemy in the Battle Against Glioblastomas: A Review. Cancers 2019, 11, 241. https://doi.org/10.3390/cancers11020241
Tomiyama A, Kobayashi T, Mori K, Ichimura K. Protein Phosphatases—A Touchy Enemy in the Battle Against Glioblastomas: A Review. Cancers. 2019; 11(2):241. https://doi.org/10.3390/cancers11020241
Chicago/Turabian StyleTomiyama, Arata, Tatsuya Kobayashi, Kentaro Mori, and Koichi Ichimura. 2019. "Protein Phosphatases—A Touchy Enemy in the Battle Against Glioblastomas: A Review" Cancers 11, no. 2: 241. https://doi.org/10.3390/cancers11020241
APA StyleTomiyama, A., Kobayashi, T., Mori, K., & Ichimura, K. (2019). Protein Phosphatases—A Touchy Enemy in the Battle Against Glioblastomas: A Review. Cancers, 11(2), 241. https://doi.org/10.3390/cancers11020241