The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endothelial Cx Expression and Its Role in Vascular Diseases
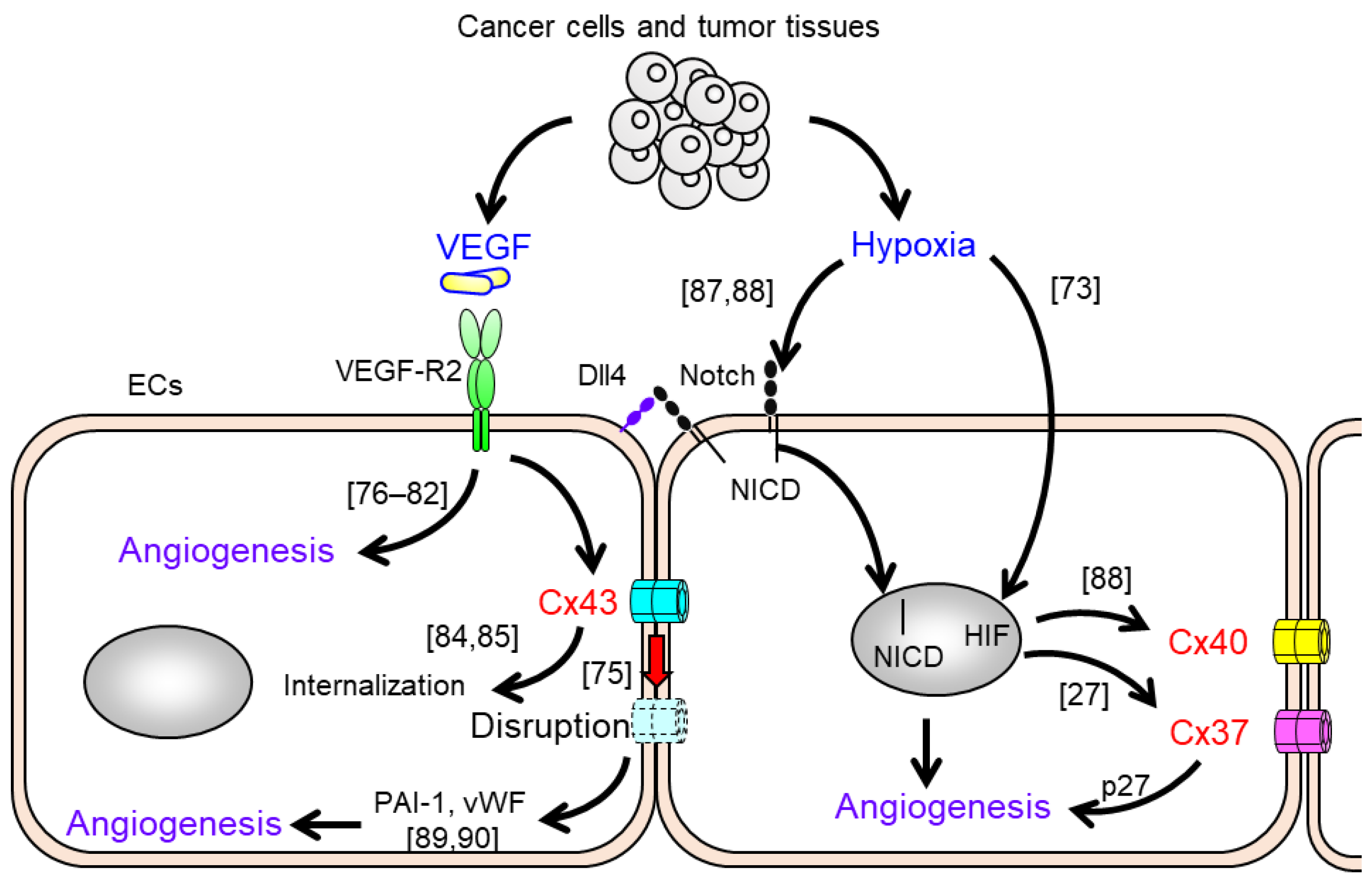
3. Alterations of Gap Junction Function and Cx Expression in Endothelial Cells in Response to Pro-Angiogenic Stimuli
4. The Impact of Endothelial Cxs on Vascular Endothelial Angiogenesis
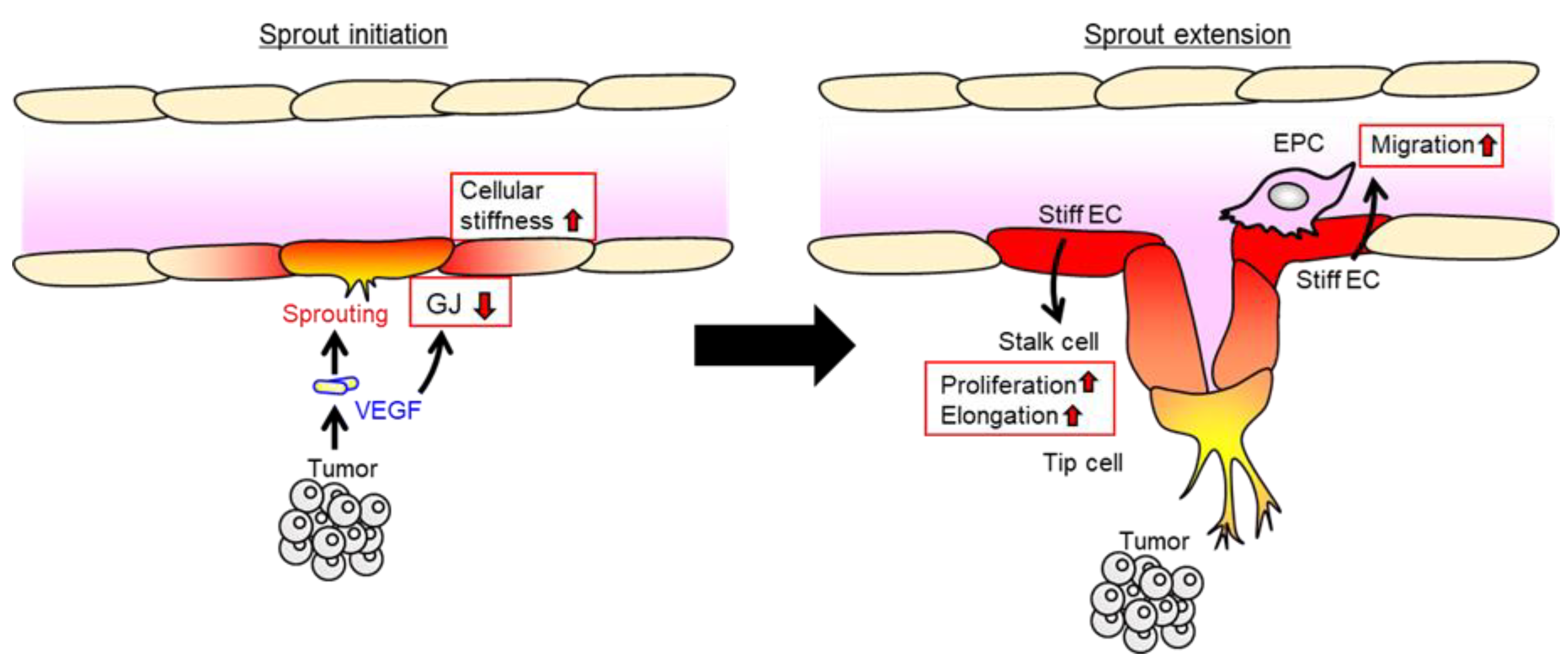
5. Endothelial Cx-Dependent Regulation of Cell Migration in Angiogenesis
6. Potential Role of Endothelial Cellular Stiffness in Cell Migration
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Patan, S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J. Neurooncol. 2000, 50, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bayless, K.J.; Johnson, G.A. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J. Vasc. Res. 2011, 48, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Keegan, A.; Hill, C.; Kumar, S.; Phillips, P.; Schor, A.; Weiss, J. Purified tumour angiogenesis factor enhances proliferation of capillary, but not aortic, endothelial cells in vitro. J. Cell Sci. 1982, 55, 261–276. [Google Scholar] [PubMed]
- Zetter, B.R. Migration of capillary endothelial cells is stimulated by tumour-derived factors. Nature 1980, 285, 41–43. [Google Scholar] [CrossRef]
- Folkman, J.; Haudenschild, C. Angiogenesis by capillary endothelial cells in culture. Trans. Ophthalmol. Soc. UK 1980, 100, 346–353. [Google Scholar]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef]
- Cao, J.; Ehling, M.; Marz, S.; Seebach, J.; Tarbashevich, K.; Sixta, T.; Pitulescu, M.E.; Werner, A.C.; Flach, B.; Montanez, E.; Raz, E.; Adams, R.H.; Schnittler, H. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat. Commun. 2017, 8, 2210. [Google Scholar] [CrossRef]
- Abedi, H.; Zachary, I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J. Biol. Chem. 1997, 272, 15442–15451. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qiu, J.; Zheng, Y.; Zhang, T.; Yin, T.; Xie, X.; Wang, G. AAMP Regulates Endothelial Cell Migration and Angiogenesis Through RhoA/Rho Kinase Signaling. Ann. Biomed. Eng. 2016, 44, 1462–1474. [Google Scholar] [CrossRef]
- Heath, V.L.; Bicknell, R. Anticancer strategies involving the vasculature. Nat. Rev. Clin. Oncol. 2009, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Hatoum, H.A.; Awada, A.H.; Ishak, R.S.; Shamseddine, A.I. Hypoxia-inducible factor in cancer angiogenesis: Structure, regulation and clinical perspectives. Crit. Rev. Oncol. Hematol. 2009, 70, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Osinsky, S.; Zavelevich, M.; Vaupel, P. Tumor hypoxia and malignant progression. Exp. Oncol. 2009, 31, 80–86. [Google Scholar] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef]
- Yeager, M.; Nicholson, B.J. Structure of gap junction intercellular channels. Curr. Opin. Struct. Biol. 1996, 6, 183–192. [Google Scholar] [CrossRef]
- Oshima, A. Structure and closure of connexin gap junction channels. FEBS Lett. 2014, 588, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Spray, D.C.; White, R.L.; Mazet, F.; Bennett, M.V. Regulation of gap junctional conductance. Am. J. Physiol. 1985, 248 Pt 2, H753–H764. [Google Scholar] [CrossRef]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.F.; Chen, L.J.; Lee, P.L.; Lee, D.Y.; Chien, S.; Chiu, J.J. Different modes of endothelial-smooth muscle cell interaction elicit differential beta-catenin phosphorylations and endothelial functions. Proc. Natl. Acad. Sci. USA 2014, 111, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Coon, B.G.; Gillis, N.; Chen, Z.; Qiu, J.; Chittenden, T.W.; Burt, J.M.; Schwartz, M.A.; Hirschi, K.K. Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun. 2017, 8, 2149. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 2007, 217, 35–41. [Google Scholar] [CrossRef]
- Johnstone, S.R.; Billaud, M.; Lohman, A.W.; Taddeo, E.P.; Isakson, B.E. Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 2012, 245, 319–332. [Google Scholar] [CrossRef]
- De Vuyst, E.; Decrock, E.; De Bock, M.; Yamasaki, H.; Naus, C.C.; Evans, W.H.; Leybaert, L. Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell 2007, 18, 34–46. [Google Scholar] [CrossRef]
- Sosinsky, G.E.; Solan, J.L.; Gaietta, G.M.; Ngan, L.; Lee, G.J.; Mackey, M.R.; Lampe, P.D. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem. J. 2007, 408, 375–385. [Google Scholar] [CrossRef] [PubMed]
- TenBroek, E.M.; Lampe, P.D.; Solan, J.L.; Reynhout, J.K.; Johnson, R.G. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 2001, 155, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.D.; Lampe, P.D. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 2002, 277, 44962–44968. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.S.; Dunn, C.A.; Carter, W.G.; Usui, M.L.; Olerud, J.E.; Lampe, P.D. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 2004, 167, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Johnstone, S.R.; Heberlein, K.R.; Rizzo, M.J.; Best, A.K.; Boitano, S.; Isakson, B.E. Site-specific connexin phosphorylation is associated with reduced heterocellular communication between smooth muscle and endothelium. J. Vasc. Res. 2010, 47, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Lin, R.; Warn-Cramer, B.J.; Lau, A.F.; Burt, J.M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 2003, 284, C511–C520. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef]
- Laing, J.G.; Beyer, E.C. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 1995, 270, 26399–26403. [Google Scholar] [CrossRef]
- Girao, H.; Pereira, P. The proteasome regulates the interaction between Cx43 and ZO-1. J. Cell Biochem. 2007, 102, 719–728. [Google Scholar] [CrossRef]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Reglero-Real, N.; Marcos-Ramiro, B.; Millan, J. Endothelial membrane reorganization during leukocyte extravasation. Cell Mol. Life Sci. 2012, 69, 3079–3099. [Google Scholar] [CrossRef]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.M.; Haudenschild, C.C.; Beyer, E.C. Gap junction messenger RNA expression by vascular wall cells. Circ. Res. 1990, 66, 1074–1080. [Google Scholar] [CrossRef]
- Kwak, B.R.; Mulhaupt, F.; Veillard, N.; Gros, D.B.; Mach, F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Gabazza, E.C.; Hayashi, T.; Suzuki, K. Connexin32 is expressed in vascular endothelial cells and participates in gap-junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 264–268. [Google Scholar] [CrossRef]
- Johnstone, S.R.; Ross, J.; Rizzo, M.J.; Straub, A.C.; Lampe, P.D.; Leitinger, N.; Isakson, B.E. Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am. J. Pathol. 2009, 175, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Suzuki, K. The Role of Gap Junction-Mediated Endothelial Cell-Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation. Int. J. Mol. Sci. 2017, 18, 2254. [Google Scholar] [CrossRef]
- van Rijen, H.V.; van Kempen, M.J.; Postma, S.; Jongsma, H.J. Tumour necrosis factor alpha alters the expression of connexin43, connexin40, and connexin37 in human umbilical vein endothelial cells. Cytokine 1998, 10, 258–264. [Google Scholar] [CrossRef]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; de Wit, C.; Kurtz, L.; Grunberger, C.; Kurtz, A.; Schweda, F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ. Res. 2007, 100, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Wong, C.; Sutter, E.; Cuhlmann, S.; Dunoyer-Geindre, S.; Mach, F.; Horrevoets, A.J.; Evans, P.C.; Krams, R.; Kwak, B.R. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell Cardiol. 2012, 53, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Pfenniger, A.; Kwak, B.R.; Delmar, M. Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovasc. Res. 2013, 99, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.E.; Paul, D.L. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ. Res. 1998, 83, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Bol, M.; Van Geyt, C.; Baert, S.; Decrock, E.; Wang, N.; De Bock, M.; Gadicherla, A.K.; Randon, C.; Evans, W.H.; Beele, H.; et al. Inhibiting connexin channels protects against cryopreservation-induced cell death in human blood vessels. Eur. J. Vasc. Endovasc. Surg. 2013, 45, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Inai, T.; Shibata, Y. Heterogeneous expression of endothelial connexin (Cx) 37, Cx40, and Cx43 in rat large veins. Anat. Sci. Int. 2009, 84, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Van Rijen, H.; van Kempen, M.J.; Analbers, L.J.; Rook, M.B.; van Ginneken, A.C.; Gros, D.; Jongsma, H.J. Gap junctions in human umbilical cord endothelial cells contain multiple connexins. Am. J. Physiol. 1997, 272 Pt 1, C117–C130. [Google Scholar] [CrossRef]
- Okamoto, T.; Akita, N.; Kawamoto, E.; Hayashi, T.; Suzuki, K.; Shimaoka, M. Endothelial connexin32 enhances angiogenesis by positively regulating tube formation and cell migration. Exp. Cell Res. 2014, 321, 133–141. [Google Scholar] [CrossRef]
- Ebong, E.E.; Kim, S.; DePaola, N. Flow regulates intercellular communication in HAEC by assembling functional Cx40 and Cx37 gap junctional channels. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2015–H2023. [Google Scholar] [CrossRef]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Akita, N.; Yoshida, K.; Hayashi, T.; Suzuki, K. Connexin32 protects against vascular inflammation by modulating inflammatory cytokine expression by endothelial cells. Exp. Cell Res. 2011, 317, 348–355. [Google Scholar] [CrossRef]
- Bolon, M.L.; Kidder, G.M.; Simon, A.M.; Tyml, K. Lipopolysaccharide reduces electrical coupling in microvascular endothelial cells by targeting connexin40 in a tyrosine-, ERK1/2-, PKA-, and PKC-dependent manner. J. Cell Physiol. 2007, 211, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Lidington, D.; Tyml, K.; Ouellette, Y. Lipopolysaccharide-induced reductions in cellular coupling correlate with tyrosine phosphorylation of connexin 43. J. Cell Physiol. 2002, 193, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Kim, N.; Gumpert, A.M.; Segretain, D.; Falk, M.M. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008, 582, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.J., 3rd; Birukova, A.A.; Beyer, E.C.; Birukov, K.G. Gap junction protein connexin43 exacerbates lung vascular permeability. PLoS ONE 2014, 9, e100931. [Google Scholar] [CrossRef]
- Chadjichristos, C.E.; Scheckenbach, K.E.; van Veen, T.A.; Richani Sarieddine, M.Z.; de Wit, C.; Yang, Z.; Roth, I.; Bacchetta, M.; Viswambharan, H.; Foglia, B.; et al. Endothelial-specific deletion of connexin40 promotes atherosclerosis by increasing CD73-dependent leukocyte adhesion. Circulation 2010, 121, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Derouette, J.P.; Wong, C.; Burnier, L.; Morel, S.; Sutter, E.; Galan, K.; Brisset, A.C.; Roth, I.; Chadjichristos, C.E.; Kwak, B.R. Molecular role of Cx37 in advanced atherosclerosis: A micro-array study. Atherosclerosis 2009, 206, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Veillard, N.; Pelli, G.; Mulhaupt, F.; James, R.W.; Chanson, M.; Mach, F. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation 2003, 107, 1033–1039. [Google Scholar] [CrossRef]
- Haefliger, J.A.; Krattinger, N.; Martin, D.; Pedrazzini, T.; Capponi, A.; Doring, B.; Plum, A.; Charollais, A.; Willecke, K.; Meda, P. Connexin43-dependent mechanism modulates renin secretion and hypertension. J. Clin. Investig. 2006, 116, 405–413. [Google Scholar] [CrossRef]
- Yamada, Y.; Izawa, H.; Ichihara, S.; Takatsu, F.; Ishihara, H.; Hirayama, H.; Sone, T.; Tanaka, M.; Yokota, M. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N. Engl. J. Med. 2002, 347, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akita, N.; Hayashi, T.; Shimaoka, M.; Suzuki, K. Endothelial connexin 32 regulates tissue factor expression induced by inflammatory stimulation and direct cell-cell interaction with activated cells. Atherosclerosis 2014, 236, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Nagibin, V.; Egan Benova, T.; Viczenczova, C.; Szeiffova Bacova, B.; Dovinova, I.; Barancik, M.; Tribulova, N. Ageing related down-regulation of myocardial connexin-43 and up-regulation of MMP-2 may predict propensity to atrial fibrillation in experimental animals. Physiol. Res. 2016, 65 (Suppl. 1), S91–S100. [Google Scholar]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Poon, R.T.; Fan, S.T.; Wong, J. Clinical implications of circulating angiogenic factors in cancer patients. J. Clin. Oncol. 2001, 19, 1207–1225. [Google Scholar] [CrossRef] [PubMed]
- Suarez, S.; Ballmer-Hofer, K. VEGF transiently disrupts gap junctional communication in endothelial cells. J. Cell Sci. 2001, 114 Pt 6, 1229–1235. [Google Scholar]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Brown, L.F.; Yeo, K.T.; Berse, B.; Yeo, T.K.; Senger, D.R.; Dvorak, H.F.; van de Water, L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 1992, 176, 1375–1379. [Google Scholar] [CrossRef]
- Maniscalco, W.M.; Watkins, R.H.; Finkelstein, J.N.; Campbell, M.H. Vascular endothelial growth factor mRNA increases in alveolar epithelial cells during recovery from oxygen injury. Am. J. Respir. Cell Mol. Biol. 1995, 13, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Hollinger, L.A.; Wolpert, E.B.; Gardner, T.W. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 1999, 274, 23463–23467. [Google Scholar] [CrossRef] [PubMed]
- Nimlamool, W.; Andrews, R.M.; Falk, M.M. Connexin43 phosphorylation by PKC and MAPK signals VEGF-mediated gap junction internalization. Mol. Biol. Cell 2015, 26, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D. The vascular endothelial growth factor-induced disruption of gap junctions is relayed by an autocrine communication via ATP release in coronary capillary endothelium. Ann. N. Y. Acad. Sci. 2004, 1030, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S.; Meda, P. Basic fibroblast growth factor increases junctional communication and connexin 43 expression in microvascular endothelial cells. J. Cell Physiol. 1992, 153, 196–205. [Google Scholar] [CrossRef]
- Favre, C.J.; Mancuso, M.; Maas, K.; McLean, J.W.; Baluk, P.; McDonald, D.M. Expression of genes involved in vascular development and angiogenesis in endothelial cells of adult lung. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1917–H1938. [Google Scholar] [CrossRef]
- Lanner, F.; Lee, K.L.; Ortega, G.C.; Sohl, M.; Li, X.; Jin, S.; Hansson, E.M.; Claesson-Welsh, L.; Poellinger, L.; Lendahl, U.; Farnebo, F. Hypoxia-induced arterial differentiation requires adrenomedullin and notch signaling. Stem Cells Dev. 2013, 22, 1360–1369. [Google Scholar] [CrossRef]
- Bacharach, E.; Itin, A.; Keshet, E. In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc. Natl. Acad. Sci. USA 1992, 89, 10686–10690. [Google Scholar] [CrossRef]
- Xu, H.; Cao, Y.; Yang, X.; Cai, P.; Kang, L.; Zhu, X.; Luo, H.; Lu, L.; Wei, L.; Bai, X.; et al. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood 2017, 130, 11–22. [Google Scholar] [CrossRef]
- Wang, W.K.; Chen, M.C.; Leong, H.F.; Kuo, Y.L.; Kuo, C.Y.; Lee, C.H. Connexin 43 suppresses tumor angiogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1alpha. Int. J. Mol. Sci. 2014, 16, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, R.C.; Yamada, K.A.; Kleber, A.G.; Saffitz, J.E. Autocrine regulation of myocyte Cx43 expression by VEGF. Circ. Res. 2002, 90, 671–677. [Google Scholar] [CrossRef]
- Fan, X.; Teng, Y.; Ye, Z.; Zhou, Y.; Tan, W.S. The effect of gap junction-mediated transfer of miR-200b on osteogenesis and angiogenesis in a co-culture of MSCs and HUVECs. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel Anjo, S.; Manadas, B.; P G Sluijter, J.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Kawamoto, E.; Gaowa, A.; Okamoto, T.; Park, E.J. Connexins and Integrins in Exosomes. Cancers (Basel) 2019, 11, 106. [Google Scholar] [CrossRef]
- Thuringer, D.; Jego, G.; Berthenet, K.; Hammann, A.; Solary, E.; Garrido, C. Gap junction-mediated transfer of miR-145-5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget 2016, 7, 28160–28168. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.Y.; Xie, L.J.; Linask, K.L.; Zhang, C.; Zhao, X.Q.; Yang, Y.; Zhou, G.M.; Wu, Y.J.; Marquez-Rosado, L.; McElhinney, D.B.; et al. Evaluating the role of connexin43 in congenital heart disease: Screening for mutations in patients with outflow tract anomalies and the analysis of knock-in mouse models. J. Cardiovasc. Dis. Res. 2011, 2, 206–212. [Google Scholar] [CrossRef]
- Ya, J.; Erdtsieck-Ernste, E.B.; de Boer, P.A.; van Kempen, M.J.; Jongsma, H.; Gros, D.; Moorman, A.F.; Lamers, W.H. Heart defects in connexin43-deficient mice. Circ. Res. 1998, 82, 360–366. [Google Scholar] [CrossRef]
- Gu, H.; Smith, F.C.; Taffet, S.M.; Delmar, M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003, 93, 201–206. [Google Scholar] [CrossRef]
- Simon, A.M.; McWhorter, A.R. Vascular abnormalities in mice lacking the endothelial gap junction proteins connexin37 and connexin40. Dev. Biol. 2002, 251, 206–220. [Google Scholar] [CrossRef]
- Haefliger, J.A.; Allagnat, F.; Hamard, L.; Le Gal, L.; Meda, P.; Nardelli-Haefliger, D.; Genot, E.; Alonso, F. Targeting Cx40 (Connexin40) Expression or Function Reduces Angiogenesis in the Developing Mouse Retina. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1872–H1881. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Kung, C.I.; Tseng, Y.Y.; Lin, Y.C.; Chen, C.H.; Tsai, C.H.; Yeh, H.I. Activation of endothelial cells to pathological status by down-regulation of connexin43. Cardiovasc. Res. 2008, 79, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Escue, R.; Manna, J.; Adebiyi, A.; Parthasarathi, K. Changes in endothelial connexin 43 expression inversely correlate with microvessel permeability and VE-cadherin expression in endotoxin-challenged lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L584–L592. [Google Scholar] [CrossRef] [PubMed]
- Gartner, C.; Ziegelhoffer, B.; Kostelka, M.; Stepan, H.; Mohr, F.W.; Dhein, S. Knock-down of endothelial connexins impairs angiogenesis. Pharmacol. Res. 2012, 65, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Pan, S.; Beningo, K.; Hupe, D. A permanent human cell line (EA.hy926) preserves the characteristics of endothelin converting enzyme from primary human umbilical vein endothelial cells. Life Sci. 1995, 56, 2331–2341. [Google Scholar] [CrossRef]
- Johnson, T.L.; Nerem, R.M. Endothelial connexin 37, connexin 40, and connexin 43 respond uniquely to substrate and shear stress. Endothelium 2007, 14, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kliche, K.; Jeggle, P.; Pavenstadt, H.; Oberleithner, H. Role of cellular mechanics in the function and life span of vascular endothelium. Pflugers Arch. 2011, 462, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Stapor, P.; Wang, X.; Goveia, J.; Moens, S.; Carmeliet, P. Angiogenesis revisited—Role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 2014, 127 Pt 20, 4331–4341. [Google Scholar] [CrossRef]
- Simpson, K.J.; Selfors, L.M.; Bui, J.; Reynolds, A.; Leake, D.; Khvorova, A.; Brugge, J.S. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 2008, 10, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Pepper, M.S.; Gros, D.B.; Meda, P. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol. Biol. Cell 2001, 12, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Su, C.H.; Wu, Y.J.; Li, J.Y.; Tseng, Y.M.; Lin, Y.C.; Hsieh, C.L.; Tsai, C.H.; Yeh, H.I. Reduction of connexin43 in human endothelial progenitor cells impairs the angiogenic potential. Angiogenesis 2013, 16, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. Biochim. Biophys. Acta 2013, 1828, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys. Acta 2012, 1818, 1993–2001. [Google Scholar] [CrossRef]
- Giepmans, B.N. Role of connexin43-interacting proteins at gap junctions. Adv. Cardiol. 2006, 42, 41–56. [Google Scholar]
- Theiss, C.; Meller, K. Microinjected anti-actin antibodies decrease gap junctional intercellular commmunication in cultured astrocytes. Exp. Cell Res. 2002, 281, 197–204. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef]
- Wei, C.J.; Francis, R.; Xu, X.; Lo, C.W. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J. Biol. Chem. 2005, 280, 19925–19936. [Google Scholar] [CrossRef]
- Vitale, M.L.; Akpovi, C.D.; Pelletier, R.M. Cortactin/tyrosine-phosphorylated cortactin interaction with connexin 43 in mouse seminiferous tubules. Microsc. Res. Tech. 2009, 72, 856–867. [Google Scholar] [CrossRef]
- Xu, X.; Li, W.E.; Huang, G.Y.; Meyer, R.; Chen, T.; Luo, Y.; Thomas, M.P.; Radice, G.L.; Lo, C.W. Modulation of mouse neural crest cell motility by N-cadherin and connexin 43 gap junctions. J. Cell Biol. 2001, 154, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Francis, R.; Wei, C.J.; Linask, K.L.; Lo, C.W. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development 2006, 133, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Kardash, E.; Reichman-Fried, M.; Maitre, J.L.; Boldajipour, B.; Papusheva, E.; Messerschmidt, E.M.; Heisenberg, C.P.; Raz, E. A role for Rho GTPases and cell-cell adhesion in single-cell motility in vivo. Nat. Cell Biol. 2010, 12, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.Y.; Zhao, X.Q.; Francis, R.J.; Huang, G.Y.; Mably, J.D.; Lo, C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development 2009, 136, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Hordijk, P.L. Cell-stiffness-induced mechanosignaling—A key driver of leukocyte transendothelial migration. J. Cell Sci. 2015, 128, 2221–2230. [Google Scholar] [CrossRef]
- Stroka, K.M.; Aranda-Espinoza, H. Effects of Morphology vs. Cell-Cell Interactions on Endothelial Cell Stiffness. Cell Mol. Bioeng. 2011, 4, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Potempa, S.; Eichholtz, T.; Ridley, A.J. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 2001, 114 Pt 7, 1343–1355. [Google Scholar]
- Wang, N.; Tolic-Norrelykke, I.M.; Chen, J.; Mijailovich, S.M.; Butler, J.P.; Fredberg, J.J.; Stamenovic, D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am. J. Physiol. Cell Physiol. 2002, 282, C606–C616. [Google Scholar] [CrossRef]
- Okamoto, T.; Kawamoto, E.; Takagi, Y.; Akita, N.; Hayashi, T.; Park, E.J.; Suzuki, K.; Shimaoka, M. Gap junction-mediated regulation of endothelial cellular stiffness. Sci. Rep. 2017, 7, 6134. [Google Scholar] [CrossRef]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Oakes, P.W.; Patel, D.C.; Morin, N.A.; Zitterbart, D.P.; Fabry, B.; Reichner, J.S.; Tang, J.X. Neutrophil morphology and migration are affected by substrate elasticity. Blood 2009, 114, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, L.; van der Stoel, M.; Rianna, C.; van Stalborch, A.M.; de Ligt, A.; Hoogenboezem, M.; Tol, S.; van Rijssel, J.; Szulcek, R.; Bogaard, H.J.; et al. Stiffness-Induced Endothelial DLC-1 Expression Forces Leukocyte Spreading through Stabilization of the ICAM-1 Adhesome. Cell Rep. 2018, 24, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Stroka, K.M.; Aranda-Espinoza, H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood 2011, 118, 1632–1640. [Google Scholar] [CrossRef]
- Schaefer, A.; Te Riet, J.; Ritz, K.; Hoogenboezem, M.; Anthony, E.C.; Mul, F.P.; de Vries, C.J.; Daemen, M.J.; Figdor, C.G.; van Buul, J.D.; et al. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J. Cell Sci. 2014, 127 Pt 20, 4470–4482. [Google Scholar] [CrossRef]
- Sack, K.D.; Teran, M.; Nugent, M.A. Extracellular Matrix Stiffness Controls VEGF Signaling and Processing in Endothelial Cells. J. Cell Physiol. 2016, 231, 2026–2039. [Google Scholar] [CrossRef]
- Browning, M.B.; Guiza, V.; Russell, B.; Rivera, J.; Cereceres, S.; Hook, M.; Hahn, M.S.; Cosgriff-Hernandez, E.M. Endothelial cell response to chemical, biological, and physical cues in bioactive hydrogels. Tissue Eng. Part A 2014, 20, 3130–3141. [Google Scholar] [CrossRef]
- Frye, M.; Taddei, A.; Dierkes, C.; Martinez-Corral, I.; Fielden, M.; Ortsater, H.; Kazenwadel, J.; Calado, D.P.; Ostergaard, P.; Salminen, M.; et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat. Commun. 2018, 9, 1511. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamoto, T.; Usuda, H.; Tanaka, T.; Wada, K.; Shimaoka, M. The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis. Cancers 2019, 11, 237. https://doi.org/10.3390/cancers11020237
Okamoto T, Usuda H, Tanaka T, Wada K, Shimaoka M. The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis. Cancers. 2019; 11(2):237. https://doi.org/10.3390/cancers11020237
Chicago/Turabian StyleOkamoto, Takayuki, Haruki Usuda, Tetsuya Tanaka, Koichiro Wada, and Motomu Shimaoka. 2019. "The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis" Cancers 11, no. 2: 237. https://doi.org/10.3390/cancers11020237
APA StyleOkamoto, T., Usuda, H., Tanaka, T., Wada, K., & Shimaoka, M. (2019). The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis. Cancers, 11(2), 237. https://doi.org/10.3390/cancers11020237