Novel Ran-RCC1 Inhibitory Peptide-Loaded Nanoparticles Have Anti-Cancer Efficacy In Vitro and In Vivo

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
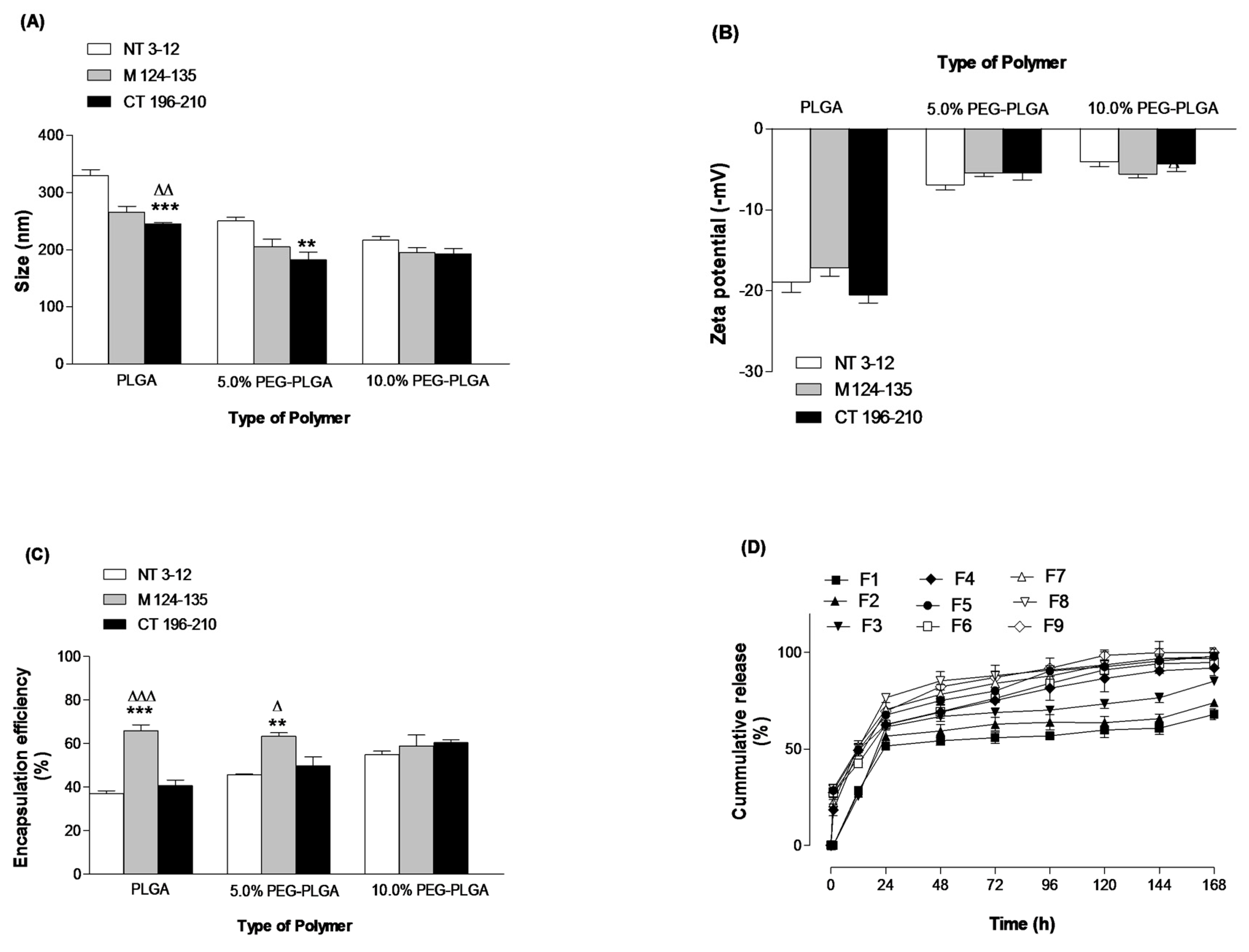
2.1. Formulation and Characterization of Peptide-Loaded NPs
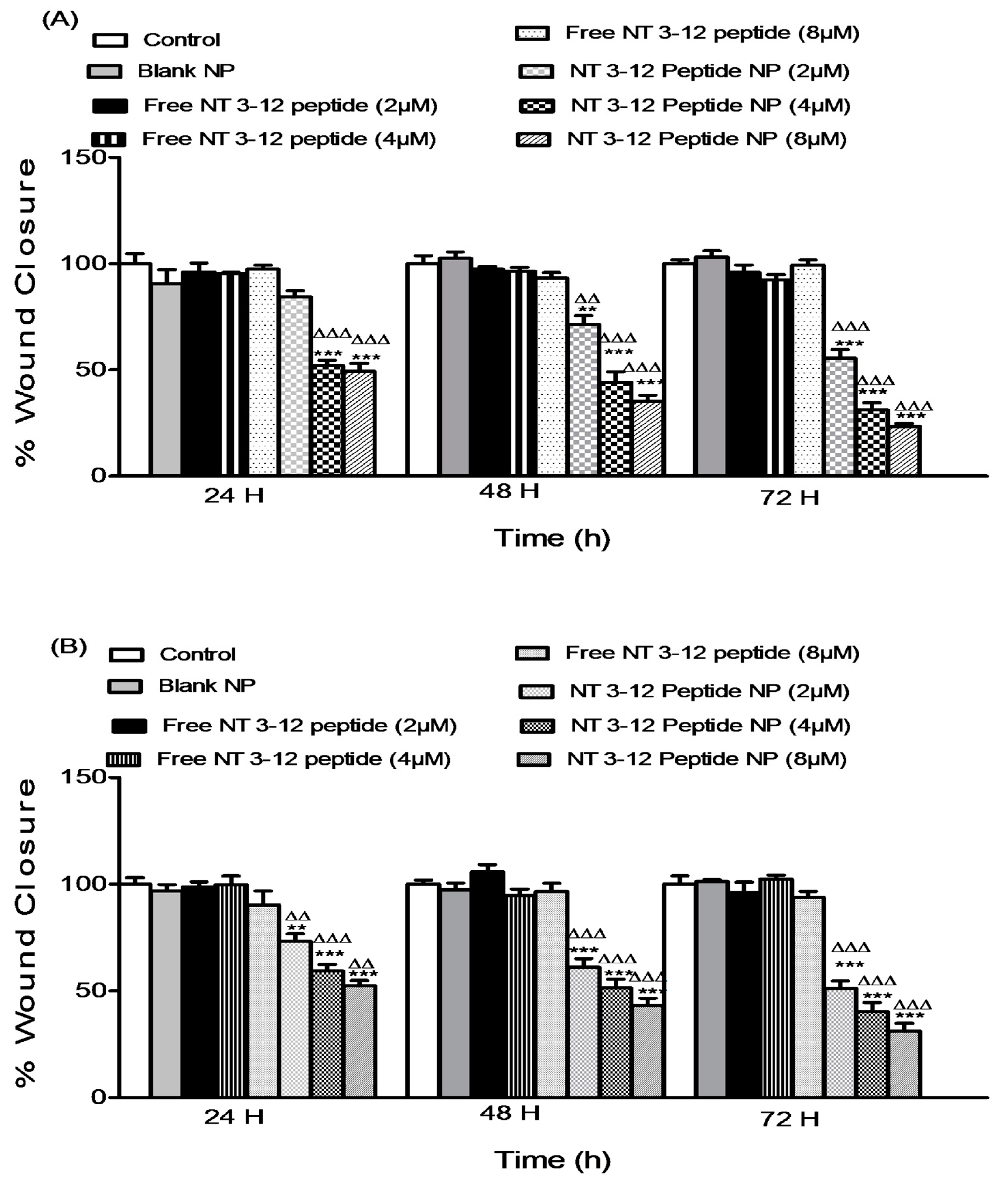
2.2. In Vitro Antitumor Activity
2.3. In Vitro Anti-Metastatic Activity
2.3.1. Cell Migration Assay
2.3.2. Cell Invasion Assay
2.3.3. Colony Formation Assay
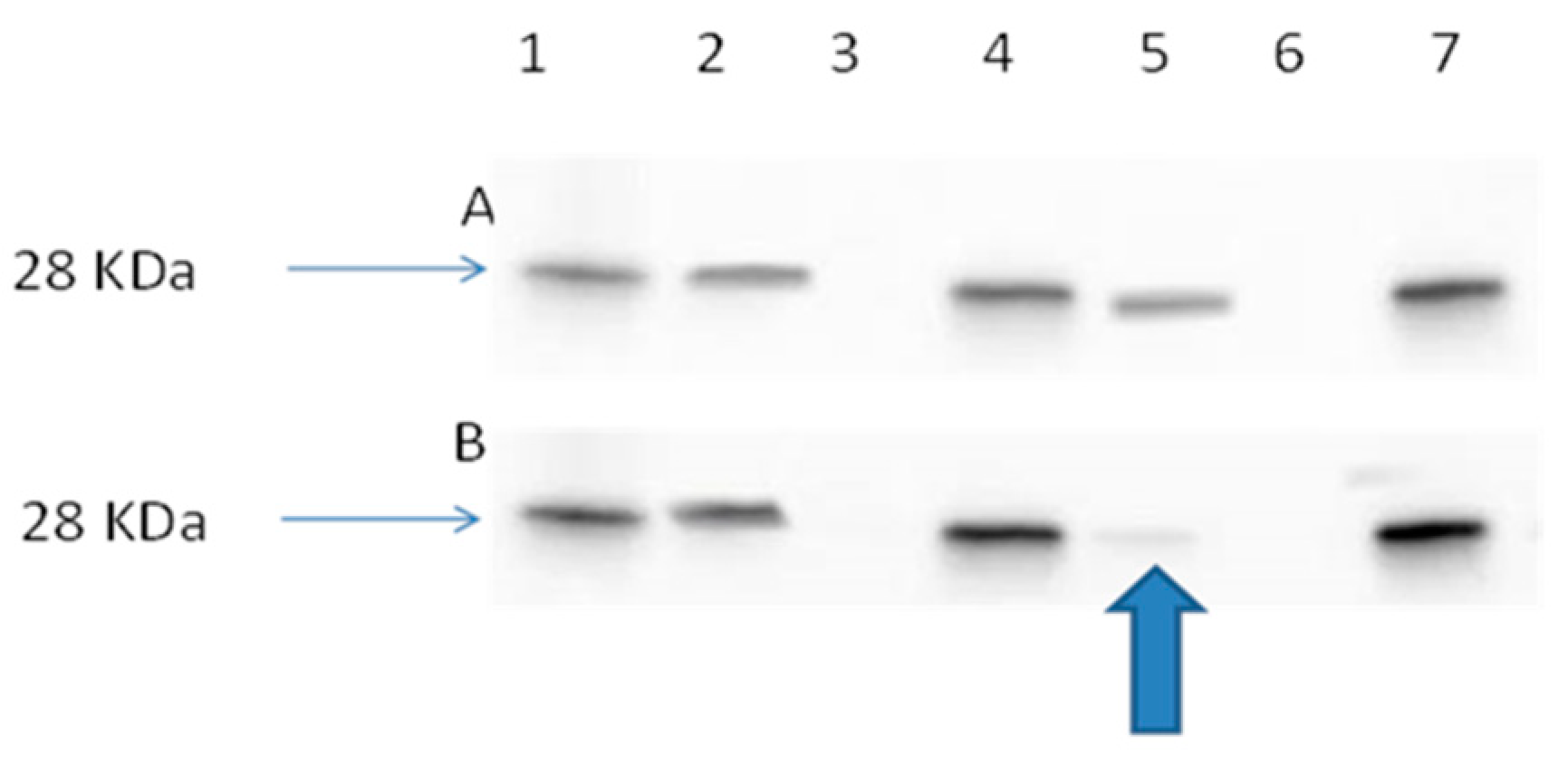
2.3.4. Ran Activation Assay
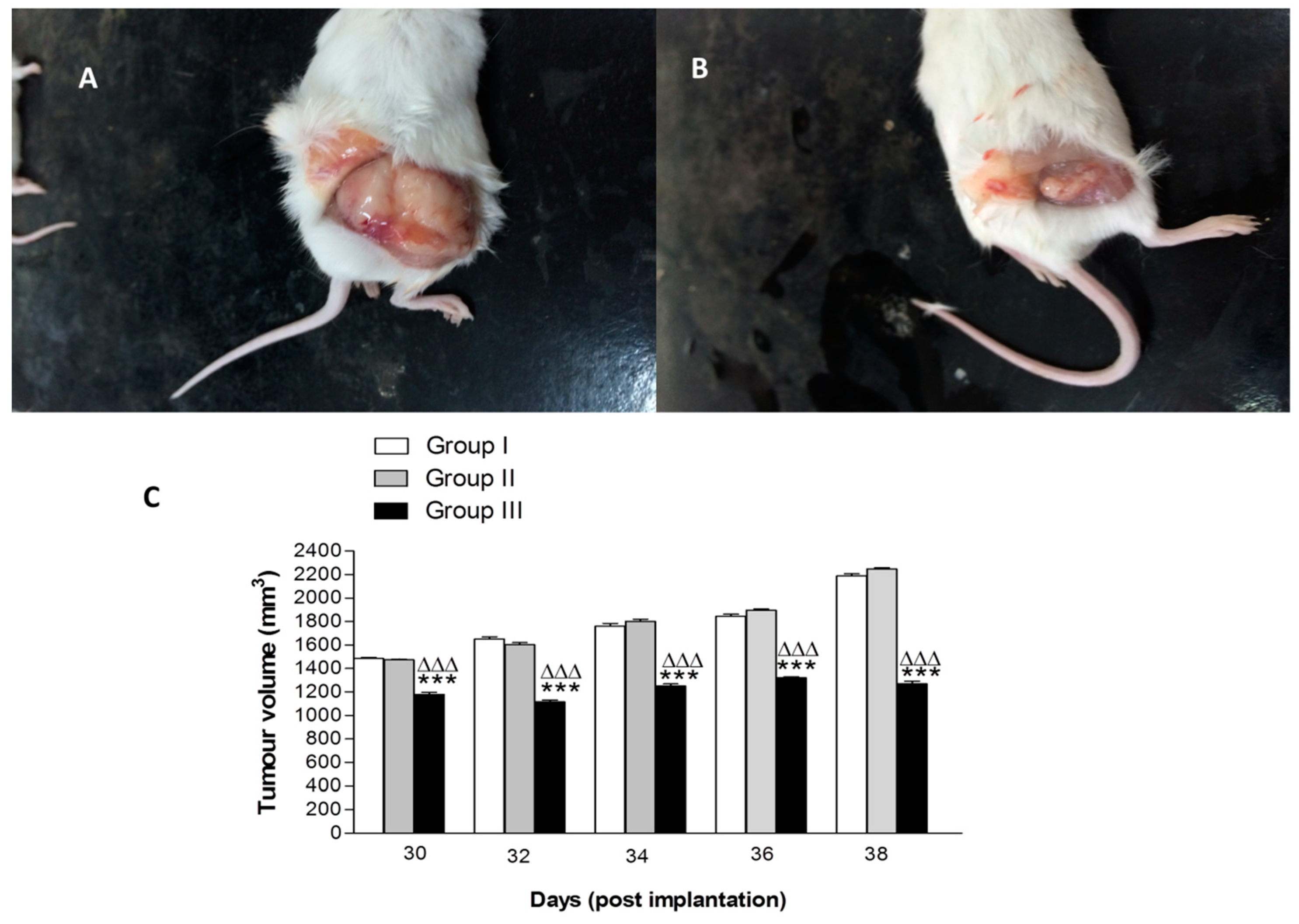
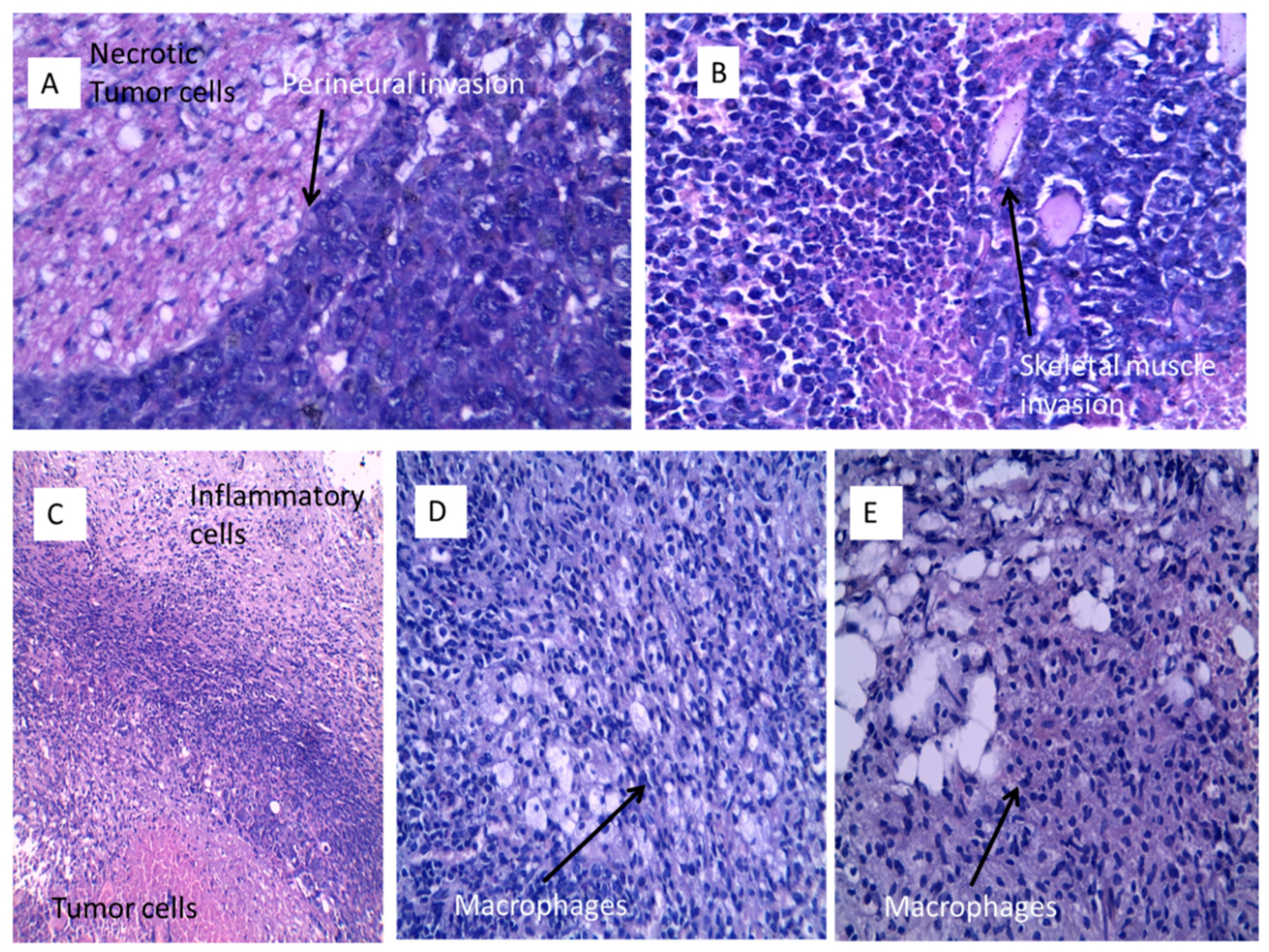
2.4. In Vivo Anti-Tumor Activity
3. Materials and Methods
3.1. Materials
3.2. Preparation of Peptide-Loaded NPs
3.3. Physico-Chemcial Characterization of Peptide-Loaded NPs
3.3.1. Particle Size and Zeta Potential
3.3.2. Particle Surface Morphology
3.3.3. Determination of Peptide Loading and Encapsulation Efficiency
3.3.4. In Vitro Release Studies
3.4. Cell Culture
3.5. In Vitro Antitumor Assay
3.6. Migration Assay
3.7. Invasion Assay
3.8. Colony Formation Assay
3.9. Ran Activation Assay
3.10. In Vivo Antitumor Activity
3.10.1. Animal Groups and Treatment Protocol
3.10.2. Tumor Volume (V) and Percentage Tumor Growth Inhibition (% TGI)
3.10.3. Processing of Tumor Tissue Samples
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.; Zhang, C. Spatial and temporal coordination of mitosis by Ran GTPase. Nat. Rev. Mol. Cell Biol. 2008, 9, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.K.; Wang, J.; Pereira, R.; Rojas, K.S.; Peng, X.; Feng, Q.; Cerione, R.A.; Wilson, K.F. Activation of the Ran GTPase is subject to growth factor regulation and can give rise to cellular transformation. J. Biol. Chem. 2010, 285, 5815–5826. [Google Scholar] [CrossRef]
- Abe, H.; Kamai, T.; Shirataki, H.; Oyama, T.; Arai, K.; Yoshida, K. High expression of Ran GTPase is associated with local invasion and metastasis of human clear cell renal cell carcinoma. Int. J. Cancer 2008, 122, 2391–2397. [Google Scholar] [CrossRef]
- Kurisetty, V.V.; Johnston, P.G.; Johnston, N.; Erwin, P.; Crowe, P.; Fernig, D.G.; Campbell, F.C.; Anderson, I.P.; Rudland, P.S.; El-Tanani, M.K. RAN GTPase is an effector of the invasive/metastatic phenotype induced by osteopontin. Oncogene 2008, 27, 7139–7149. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; Chan, K.K.; Platt-Higgins, A.; Dakir, E.-H.; Matchett, K.B.; Haggag, Y.A.; Jithesh, P.V.; Habib, T.; Faheem, A.; Dean, F.A.; et al. Ran GTPase promotes cancer progression via Met receptormediated downstream signaling. Oncotarget 2016, 7, 75854–75864. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; Chan, K.K.; Grills, C.; Murray, J.T.; Platt-Higgins, A.; Eldin, O.S.; O’Byrne, K.; Janne, P.; Fennell, D.A.; Johnston, P.G.; et al. Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways. Clin. Cancer Res. 2012, 18, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Lee, C.W.; Altieri, D.C. Tumor cell dependence on Ran-GTP-directed mitosis. Cancer Res. 2008, 68, 1826–1833. [Google Scholar] [CrossRef]
- Vetter, I.R.; Nowak, C.; Nishimoto, T.; Kuhlmann, J.; Wittinghofer, A. Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: Implications for nuclear transport. Nature 1999, 398, 39–46. [Google Scholar] [CrossRef]
- Scheffzek, K.; Klebe, C.; Fritz-Wolf, K.; Kabsch, W.; Wittinghofer, A. Crystal structure of the nuclear Ras-related protein Ran in its GDP-bound form. Nature 1995, 374, 378–381. [Google Scholar] [CrossRef]
- Bischoff, F.R.; Ponstingl, H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 1991, 354, 80–82. [Google Scholar] [CrossRef]
- Klebe, C.; Prinz, H.; Wittinghofer, A.; Goody, R.S. The kinetic mechanism of Ran--nucleotide exchange catalyzed by RCC1. Biochemistry 1995, 34, 12543–12552. [Google Scholar] [CrossRef] [PubMed]
- Nemergut, M.E.; Mizzen, C.A.; Stukenberg, T.; Allis, C.D.; Macara, I.G. Chromatin docking and exchange activity enhancement of RCC1 by histones H2A and H2B. Science 2001, 292, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Haggag, Y.A.; Matchett, K.B.; Dakir El, H.; Buchanan, P.; Osman, M.A.; Elgizawy, S.A.; El-Tanani, M.; Faheem, A.M.; McCarron, P.A. Nano-encapsulation of a novel anti-Ran-GTPase peptide for blockade of regulator of chromosome condensation 1 (RCC1) function in MDA-MB-231 breast cancer cells. Int. J. Pharm. 2017, 521, 40–53. [Google Scholar] [CrossRef]
- Brigger, I.; Dubernet, C.; Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug Deliv. Rev. 2002, 54, 631–651. [Google Scholar] [CrossRef]
- Crawford, E.D.; Phillips, J.M. Six-month gonadotropin releasing hormone (GnRH) agonist depots provide efficacy, safety, convenience, and comfort. Cancer Manag. Res. 2011, 3, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Borghouts, C.; Kunz, C.; Groner, B. Current strategies for the development of peptide-based anti-cancer therapeutics. J. Pept. Sci. 2005, 11, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Lev, D.C.; Kim, L.S.; Melnikova, V.; Ruiz, M.; Ananthaswamy, H.N.; Price, J.E. Dual blockade of EGFR and ERK1/2 phosphorylation potentiates growth inhibition of breast cancer cells. Br. J. Cancer 2004, 91, 795–802. [Google Scholar] [CrossRef]
- Antošová, Z.; Macková, M.; Král, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef]
- Haggag, Y.A.; Faheem, A.M. Evaluation of nano spray drying as a method for drying and formulation of therapeutic peptides and proteins. Front. Pharmacol. 2015, 6, 140. [Google Scholar] [CrossRef]
- Mohan, A.; McClements, D.J.; Udenigwe, C.C. Encapsulation of bioactive whey peptides in soy lecithin-derived nanoliposomes: Influence of peptide molecular weight. Food Chem. 2016, 213, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Derman, S.; Mustafaeva, Z.A.; Abamor, E.S.; Bagirova, M.; Allahverdiyev, A. Preparation, characterization and immunological evaluation: Canine parvovirus synthetic peptide loaded PLGA nanoparticles. J. Biomed. Sci. 2015, 22, 89. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Haggag, Y.; Abdel-Wahab, Y.; Ojo, O.; Osman, M.; El-Gizawy, S.; El-Tanani, M.; Faheem, A.; McCarron, P. Preparation and in vivo evaluation of insulin-loaded biodegradable nanoparticles prepared from diblock copolymers of PLGA and PEG. Int. J. Pharm. 2016, 499, 236–246. [Google Scholar] [CrossRef]
- Danhier, D.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Vasir, J.K.; Labhasetwar, V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv. Drug Deliv. Rev. 2007, 59, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.Z.; Zeng, Z.W.; Zhou, G.L.; Wang, J.J.; Li, F.Z.; Wang, A.M. Recent advances in PEG-PLA block copolymer nanoparticles. Int. J. Nanomedicine 2010, 5, 1057–1065. [Google Scholar] [CrossRef]
- Moore, W.; Zhang, C.; Clarke, P.R. Targeting of RCC1 to chromosomes is required for proper mitotic spindle assembly in human cells. Curr. Biol. 2002, 12, 1442–1447. [Google Scholar] [CrossRef]
- Chen, T.; Muratore, T.L.; Schaner-Tooley, C.E.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G. N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat. Cell Biol. 2007, 9, 596–603. [Google Scholar] [CrossRef]
- Palmer, T.D.; Ashby, W.J.; Lewis, J.D.; Zijlstra, A. Targeting tumor cell motility to prevent metastasis. Adv. Drug Deliv. Rev. 2011, 63, 568–581. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J. Colloid Interface Sci. 2010, 351, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Nascimento, K.A.; Maciel, M.C.; Pinheiro, M.T.; Sousa, P.R.; Ferreira, S.C.; Azevedo, A.P.; Guerra, R.N.; Nascimento, F.R. Sunflower seed oil-enriched product can inhibit Ehrlich solid tumor growth in mice. Chemotherapy 2006, 52, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Ferraz-de-Paula, V.; Pinheiro, M.L.; Ribeiro, A.; Quinteiro-Filho, W.M.; Rone, M.B.; Martinez-Arguelles, D.B.; Dagli, M.L.Z.; Papadopoulos, V.; Palermo-Neto, J. Translocator protein (18 kDa) mediates the pro-growth effects of diazepam on Ehrlich tumor cells in vivo. Eur. J. Pharmacol. 2010, 626, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, N.; Tachibana, T.; Matsubae, M.; Yoneda, Y. A karyophilic protein forms a stable complex with cytoplasmic components prior to nuclear pore binding. J Biol. Chem. 1995, 270, 8559–8565. [Google Scholar] [CrossRef] [PubMed]
- Kotera, I.; Sekimoto, T.; Miyamoto, Y.; Saiwaki, T.; Nagoshi, E.; Sakagami, H.; Kondo, H.; Yoneda, Y. Importin α transports CaMKIV to the nucleus without utilizing importin β. EMBO J. 2005, 24, 942–951. [Google Scholar] [CrossRef]
- De Luca, A.; Mangiacasale, R.; Severino, A.; Malquori, L.; Baldi, A.; Palena, A.; Mileo, A.M.; Lavia, P.; Paggi, M.G. E1A deregulates the centrosome cycle in a Ran GTPase-dependent manner. Cancer Res. 2003, 63, 1430–1437. [Google Scholar] [PubMed]
- van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Haggag, Y.A.; Faheem, A.M.; Tambuwala, M.M.; Osman, M.A.; El-Gizawy, S.A.; O’Hagan, B.; Irwin, N.; McCarron, P.A. Effect of poly(ethylene glycol) content and formulation parameters on particulate properties and intraperitoneal delivery of insulin from PLGA nanoparticles prepared using the double-emulsion evaporation procedure. Pharm. Dev. Technol. 2018, 23, 370–381. [Google Scholar] [CrossRef]
- Haggag, Y.A.; Osman, M.A.; El-Gizawy, S.A.; Goda, A.E.; Shamloula, M.M.; Faheem, A.M.; McCarron, P.A. Polymeric nano-encapsulation of 5-fluorouracil enhances anti-cancer activity and ameliorates side effects in solid Ehrlich Carcinoma-bearing mice. Biomed. Pharmacother. 2018, 105, 215–224. [Google Scholar] [CrossRef]
- Kumar, S.S.; Mahesh, A.; Mahadevan, S.; Mandal, A.B. Synthesis and characterization of curcumin loaded polymer/lipid based nanoparticles and evaluation of their antitumor effects on MCF-7 cells. Biochim. Biophys. Acta 2014, 1840, 1913–1922. [Google Scholar] [CrossRef]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protocols 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; Gunasekharan, V.K.; Chan, K.K.; Zhang, S.D.; Platt-Higgins, A.; Gately, K.; O’Byrne, K.; Fennell, D.A.; Johnston, P.G.; Rudland, P.S.; et al. RanGTPase: A candidate for Myc-mediated cancer progression. J. Natl. Cancer Inst. 2013, 105, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.e.-M.; Ahmed, M.M.; Khayyal, M.T.; El-Merzabani, M.M. Hyperthermic potentiation of cisplatin cytotoxicity on solid Ehrlich carcinoma. Tumori 1993, 79, 268–272. [Google Scholar] [CrossRef]
- Awara, W.M.; El-Sisi, A.E.; El-Sayad, M.E.; Goda, A.E. The potential role of cyclooxygenase-2 inhibitors in the treatment of experimentally-induced mammary tumor: Does celecoxib enhance the anti-tumor activity of doxorubicin? Pharmacol. Res. 2004, 50, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Kimler, B.F.; Estes, N.C.; Durham, F.J. Growth delay effect of combined interstitial hyperthermia and brachytherapy in a rat solid tumor model. Anticancer Res. 1989, 9, 45–47. [Google Scholar]
- Sancéau, J.; Poupon, M.F.; Delattre, O.; Sastre-Garau, X.; Wietzerbin, J. Strong inhibition of Ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene 2002, 21, 7700–7709. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula ID | Polymer Type | Encapsulated Peptide * |
|---|---|---|
| F1 | PLGA | NT 3–12 |
| F2 | PLGA | M 124–135 |
| F3 | PLGA | CT 196–210 |
| F4 | 5% PEG-PLGA | NT 3–12 |
| F5 | 5% PEG-PLGA | M 124–135 |
| F6 | 5% PEG-PLGA | CT 196–210 |
| F7 | 10% PEG-PLGA | NT 3–12 |
| F8 | 10% PEG-PLGA | M 124–135 |
| F9 | 10% PEG-PLGA | CT 196–210 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haggag, Y.A.; Matchett, K.B.; Falconer, R.A.; Isreb, M.; Jones, J.; Faheem, A.; McCarron, P.; El-Tanani, M. Novel Ran-RCC1 Inhibitory Peptide-Loaded Nanoparticles Have Anti-Cancer Efficacy In Vitro and In Vivo. Cancers 2019, 11, 222. https://doi.org/10.3390/cancers11020222
Haggag YA, Matchett KB, Falconer RA, Isreb M, Jones J, Faheem A, McCarron P, El-Tanani M. Novel Ran-RCC1 Inhibitory Peptide-Loaded Nanoparticles Have Anti-Cancer Efficacy In Vitro and In Vivo. Cancers. 2019; 11(2):222. https://doi.org/10.3390/cancers11020222
Chicago/Turabian StyleHaggag, Yusuf A., Kyle B. Matchett, Robert A. Falconer, Mohammad Isreb, Jason Jones, Ahmed Faheem, Paul McCarron, and Mohamed El-Tanani. 2019. "Novel Ran-RCC1 Inhibitory Peptide-Loaded Nanoparticles Have Anti-Cancer Efficacy In Vitro and In Vivo" Cancers 11, no. 2: 222. https://doi.org/10.3390/cancers11020222
APA StyleHaggag, Y. A., Matchett, K. B., Falconer, R. A., Isreb, M., Jones, J., Faheem, A., McCarron, P., & El-Tanani, M. (2019). Novel Ran-RCC1 Inhibitory Peptide-Loaded Nanoparticles Have Anti-Cancer Efficacy In Vitro and In Vivo. Cancers, 11(2), 222. https://doi.org/10.3390/cancers11020222