Paradoxical Roles of Oxidative Stress Response in the Digestive System before and after Carcinogenesis

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Perspective of this Review
3. Current Understanding of Oxidative Stress in the Digestive System
3.1. Oxidative Stress in Chronic Digestive Diseases
3.2. Problems in the Management of Oxidative Stress
3.2.1. How to Monitor Oxidative Stress
3.2.2. The Control of Oxidative Stress
4. Oxidative Stress in the Upper Gastrointestinal Tract
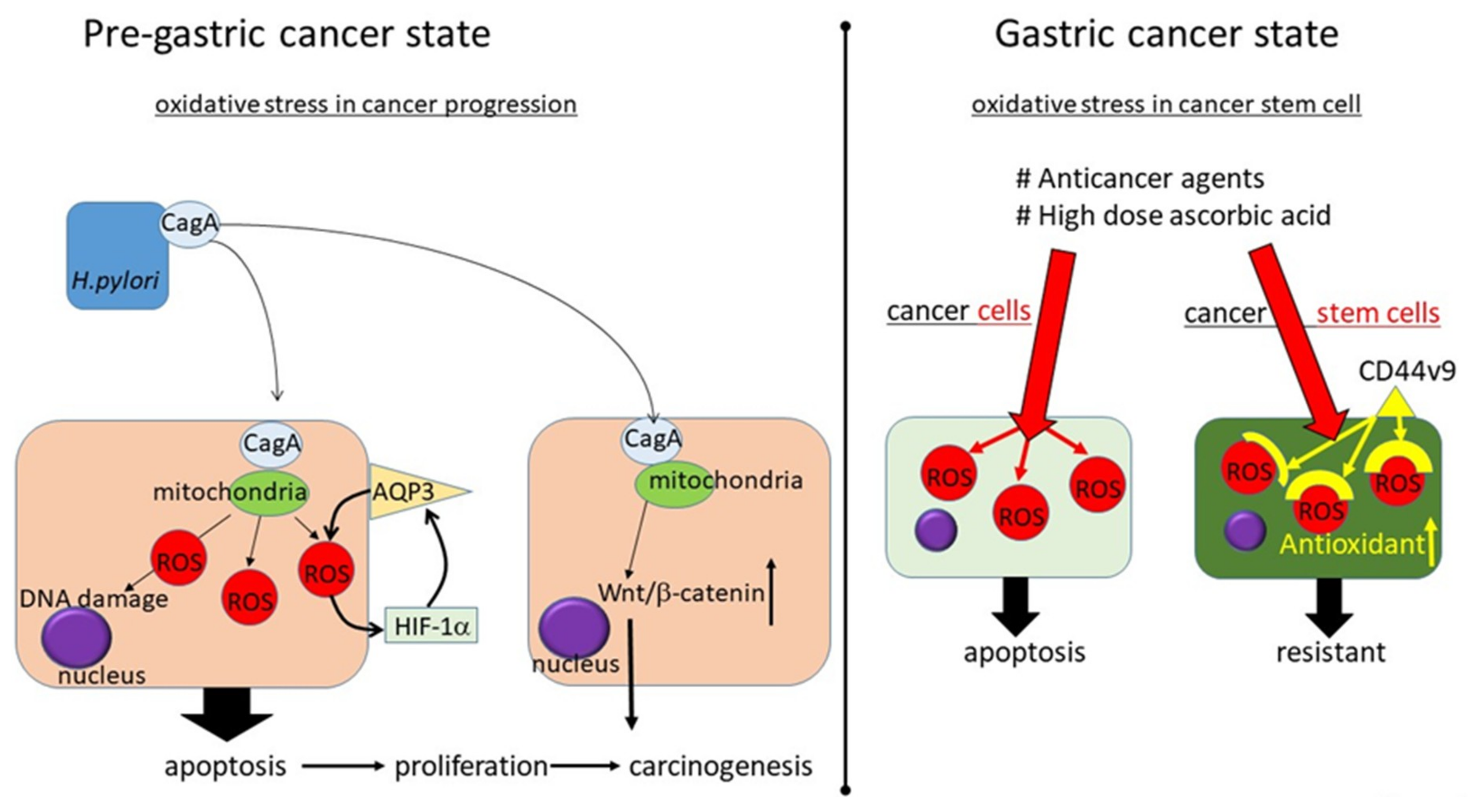
4.1. Oxidative Stress in H. pylori-Related Gastric Carcinogenesis: Before Carcinogenesis
4.2. How to Manage Oxidative Stress in Gastric Cancer: After Gastric Carcinogenesis
5. Oxidative Stress in the Colon
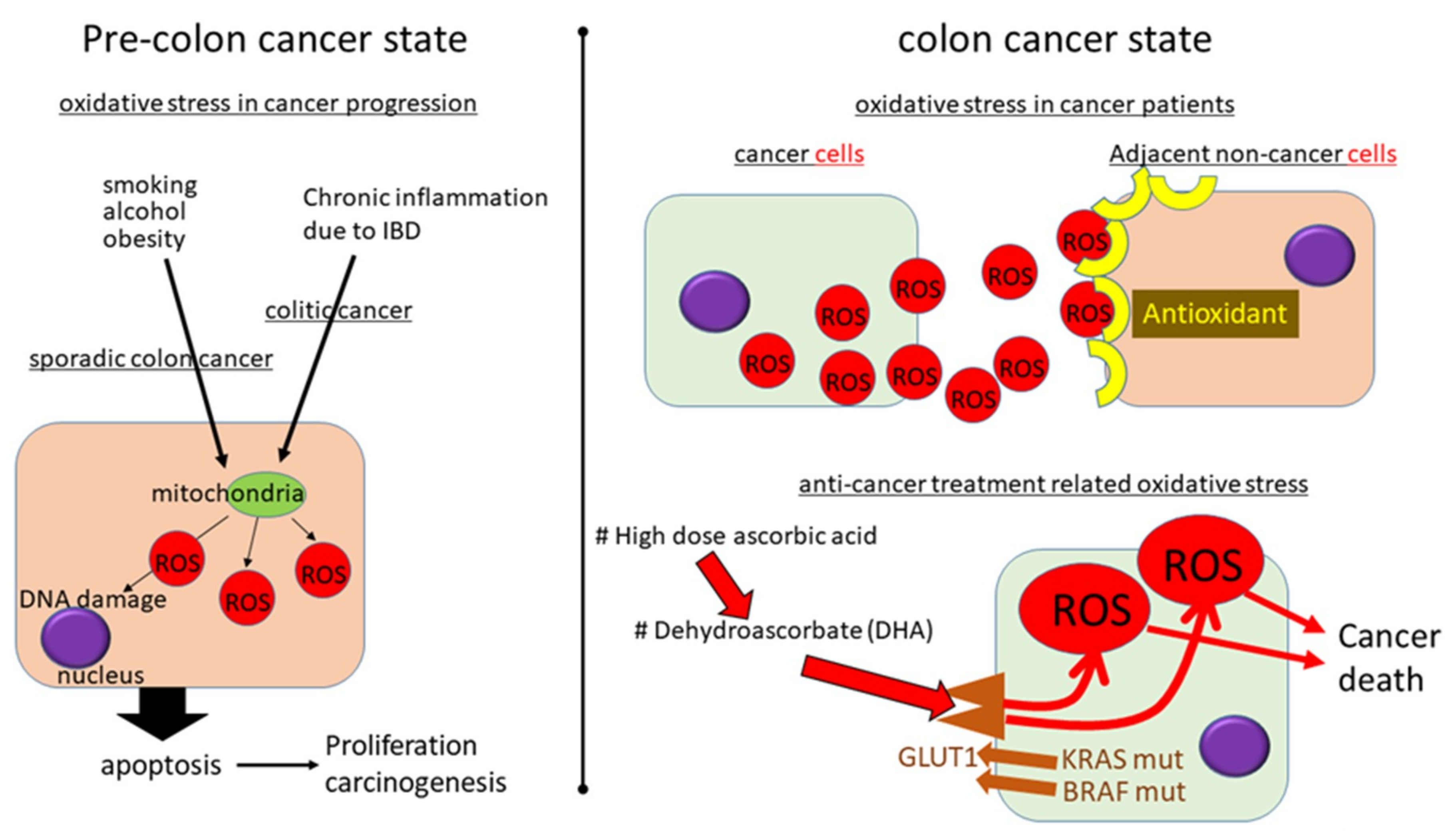
5.1. Oxidative Stress in Sporadic CRC
5.2. Oxidative Stress in Colitic Carcinogenesis
5.3. How to Manage Oxidative Stress to Prevent CRC Development: Before Carcinogenesis
5.4. How to Manage Oxidative Stress in CRC: After Carcinogenesis
6. Oxidative Stress in the Liver
6.1. Oxidative Stress in Hepatitis Virus-Related Hepatocarcinogenesis: Before Carcinogenesis
6.1.1. Oxidative Stress in HBV-Related Hepatocarcinogenesis: Before Carcinogenesis
6.1.2. Oxidative Stress in HCV-Related Hepatocarcinogenesis: Before Carcinogenesis
6.2. Oxidative Stress in NAFLD-Related Hepatocarcinogenesis: Before Carcinogenesis
6.3. How to Manage Oxidative Stress in HCC: After Hepatocarcinogenesis
7. Oxidative Stress in Pancreaticobiliary Tract Diseases
7.1. Oxidative Stress in Pancreatic Carcinogenesis: Before Carcinogenesis
7.2. Oxidative Stress in Biliary Tract Carcinogenesis: Before Carcinogenesis
7.3. How to Manage Oxidative Stress in Pancreaticobiliary Tract Cancers: After Carcinogenesis
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Pacifico, L.; Anania, C.; Martino, F.; Poggiogalle, E.; Chiarelli, F.; Arca, M.; Chiesa, C. Management of metabolic syndrome in children and adolescents. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Doycheva, I.; Watt, K.D.; Alkhouri, N. Nonalcoholic fatty liver disease in adolescents and young adults: The next frontier in the epidemic. Hepatology 2017, 65, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S20–S27. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Jaiswal, J.K. Cellular mechanisms and signals that coordinate plasma membrane repair. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int. 2009, 3, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Yongvanit, P.; Pinlaor, S.; Bartsch, H. Oxidative and nitrative DNA damage: Key events in opisthorchiasis-induced carcinogenesis. Parasitol. Int. 2012, 61, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hwang, I.; Kang, Y.N.; Choi, I.J.; Kim, D.K. Genetic characteristics of mitochondrial DNA was associated with colorectal carcinogenesis and its prognosis. PLoS ONE 2015, 10, e0118612. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. HNE as an inducer of COX-2. Free Radic. Biol. Med. 2017, 111, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.U.; Hahne, G.; Hanske, J.; Bange, T.; Bier, D.; Rademacher, C.; Hennig, S.; Grossmann, T.N. Redox Modulation of PTEN Phosphatase Activity by Hydrogen Peroxide and Bisperoxidovanadium Complexes. Angew. Chem. Int. Ed. 2015, 54, 13796–13800. [Google Scholar] [CrossRef]
- Montero, A.J.; Jassem, J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs 2011, 71, 1385–1396. [Google Scholar] [CrossRef]
- Salerno, L.; Romeo, G.; Modica, M.N.; Amata, E.; Sorrenti, V.; Barbagallo, I.; Pittala, V. Heme oxygenase-1: A new druggable target in the management of chronic and acute myeloid leukemia. Eur. J. Med. Chem. 2017, 142, 163–178. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Non-alcoholic fatty liver disease. Crit. Rev. Clin. Lab. Sci. 2011, 48, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Sezaki, T.; Tamura-Nakano, M.; Oyama, C.; Hagiwara, T.; Ishikawa, T.; Fukuda, S.; Yamada, K.; Higuchi, K.; Dohi, T.; et al. Fatty acids in a high-fat diet potentially induce gastric parietal-cell damage and metaplasia in mice. J. Gastroenterol. 2017, 52, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Vassalle, C.; Pingitore, A.; De Giuseppe, R.; Vigna, L.; Bamonti, F. Biomarkers Part II: Biomarkers to Estimate Bioefficacy of Dietary/Supplemental Antioxidants in Sport. In Antioxidants in Sport Nutrition; Lamprecht, M., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2015. [Google Scholar]
- Pingitore, A.; Lima, G.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and oxidative stress: Potential effects of antioxidant dietary strategies in sports. Nutrition 2015, 31, 916–922. [Google Scholar] [CrossRef]
- Peternelj, T.T.; Coombes, J.S. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med. 2011, 41, 1043–1069. [Google Scholar] [CrossRef]
- Taylor, P.R.; Li, B.; Dawsey, S.M.; Li, J.Y.; Yang, C.S.; Guo, W.; Blot, W.J. Prevention of esophageal cancer: The nutrition intervention trials in Linxian, China. Linxian Nutrition Intervention Trials Study Group. Cancer Res. 1994, 54, 2029s–2031s. [Google Scholar]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L., Jr.; Valanis, B.; Williams, J.H., Jr.; et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J. Natl. Cancer Inst. 1996, 88, 1550–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shu, X.O.; Li, H.; Yang, G.; Cai, H.; Ji, B.T.; Gao, J.; Gao, Y.T.; Zheng, W.; Xiang, Y.B. Vitamin intake and liver cancer risk: A report from two cohort studies in China. J. Natl. Cancer Inst. 2012, 104, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Suzuki, M.; Mori, M.; Kitahora, T.; Yokoyama, H.; Miura, S.; Hibi, T.; Ishii, H. Augmented levels of gastric mucosal leucocyte activation by infection with cagA gene-positive Helicobacter pylori. J. Gastroenterol. Hepatol. 1998, 13, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Nishikawa, M.; Higuchi, K.; Hirose, M.; Wei, Q.; Haque, A.; Sasaki, E.; Shiba, M.; Tominaga, K.; Watanabe, T.; et al. Production of reactive oxygen species in peripheral blood is increased in individuals with Helicobacter pylori infection and decreased after its eradication. Helicobacter 2006, 11, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Senda, M.; Morohashi, H.; Higashi, H.; Horio, M.; Kashiba, Y.; Nagase, L.; Sasaya, D.; Shimizu, T.; Venugopalan, N.; et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 2012, 12, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Jung, D.E.; Yu, S.S.; Lee, Y.S.; Choi, B.K.; Lee, Y.C. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget 2017, 8, 78365–78378. [Google Scholar] [CrossRef]
- Yong, X.; Tang, B.; Xiao, Y.F.; Xie, R.; Qin, Y.; Luo, G.; Hu, C.J.; Dong, H.; Yang, S.M. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/beta-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016, 374, 292–303. [Google Scholar] [CrossRef]
- Aikman, B.; de Almeida, A.; Meier-Menches, S.M.; Casini, A. Aquaporins in cancer development: Opportunities for bioinorganic chemistry to contribute novel chemical probes and therapeutic agents. Metallomics 2018, 10, 696–712. [Google Scholar] [CrossRef]
- Shen, L.; Zhu, Z.; Huang, Y.; Shu, Y.; Sun, M.; Xu, H.; Zhang, G.; Guo, R.; Wei, W.; Wu, W. Expression profile of multiple aquaporins in human gastric carcinoma and its clinical significance. Biomed. Pharmacother. 2010, 64, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, Y.; Gao, C.; Zhang, G.; You, Q.; Zhang, W.; Zhang, Z.; Wang, S.; Peng, G.; Shen, L. Helicobacter pylori infection promotes Aquaporin 3 expression via the ROS-HIF-1alpha-AQP3-ROS loop in stomach mucosa: A potential novel mechanism for cancer pathogenesis. Oncogene 2018, 37, 3549–3561. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Sasazuki, S.; Shimazu, T.; Sawada, N.; Yamaji, T.; Iwasaki, M.; Inoue, M.; Tsugane, S. Reactive oxygen species and gastric cancer risk: A large nested case-control study in Japan. Eur. J. Epidemiol. 2015, 30, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Taylor, P.R.; Fan, J.H.; Pfeiffer, R.M.; Gail, M.H.; Liang, H.; Murphy, G.A.; Dawsey, S.M.; Qiao, Y.L.; Abnet, C.C. Effects of Nutrition Intervention on Total and Cancer Mortality: 25-Year Post-trial Follow-up of the 5.25-Year Linxian Nutrition Intervention Trial. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef]
- Fang, X.; Wei, J.; He, X.; An, P.; Wang, H.; Jiang, L.; Shao, D.; Liang, H.; Li, Y.; Wang, F.; et al. Landscape of dietary factors associated with risk of gastric cancer: A systematic review and dose-response meta-analysis of prospective cohort studies. Eur. J. Cancer 2015, 51, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Blot, W.J.; Li, J.Y.; Taylor, P.R.; Guo, W.; Dawsey, S.; Wang, G.Q.; Yang, C.S.; Zheng, S.F.; Gail, M.; Li, G.Y.; et al. Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 1993, 85, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.L.; Dawsey, S.M.; Kamangar, F.; Fan, J.H.; Abnet, C.C.; Sun, X.D.; Johnson, L.L.; Gail, M.H.; Dong, Z.W.; Yu, B.; et al. Total and cancer mortality after supplementation with vitamins and minerals: Follow-up of the Linxian General Population Nutrition Intervention Trial. J. Natl. Cancer Inst. 2009, 101, 507–518. [Google Scholar] [CrossRef]
- You, X.; Ma, M.; Hou, G.; Hu, Y.; Shi, X. Gene expression and prognosis of NOX family members in gastric cancer. Onco Targets Ther. 2018, 11, 3065–3074. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Suzuki, H.; Imaeda, H.; Matsuzaki, J.; Tsugawa, H.; Nagano, O.; Asakura, K.; Saya, H.; Hibi, T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 2013, 109, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Oshima, H.; Oshima, M.; Kai, K.; Torii, R.; Masuko, T.; Baba, H.; Saya, H.; Nagano, O. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010, 101, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- O’Leary, B.R.; Houwen, F.K.; Johnson, C.L.; Allen, B.G.; Mezhir, J.J.; Berg, D.J.; Cullen, J.J.; Spitz, D.R. Pharmacological Ascorbate as an Adjuvant for Enhancing Radiation-Chemotherapy Responses in Gastric Adenocarcinoma. Radiat. Res. 2018, 189, 456–465. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Perse, M. Oxidative stress in the pathogenesis of colorectal cancer: Cause or consequence? Biomed. Res. Int. 2013, 2013, 725710. [Google Scholar] [CrossRef]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.Y.; Bostick, R.M.; Flanders, W.D.; McClellan, W.M.; Thyagarajan, B.; Gross, M.D.; Judd, S.; Goodman, M. Oxidative balance score, colorectal adenoma, and markers of oxidative stress and inflammation. Cancer Epidemiol. Prev. Biomark. 2014, 23, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Lopez, R.; Torrens-Mas, M.; Pons, D.G.; Company, M.M.; Falco, E.; Fernandez, T.; Ibarra de la Rosa, J.M.; Sastre-Serra, J.; Oliver, J.; Roca, P. Non-tumor adjacent tissue of advanced stage from CRC shows activated antioxidant response. Free Radic. Biol. Med. 2018, 126, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T. Epigenetic field for cancerization. J. Biochem. Mol. Biol. 2007, 40, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Van der Logt, E.M.; Roelofs, H.M.; Wobbes, T.; Nagengast, F.M.; Peters, W.H. High oxygen radical production in patients with sporadic colorectal cancer. Free Radic. Biol. Med. 2005, 39, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Yoshikawa, T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J. Gastroenterol. 2007, 42, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Costa Pereira, C.; Duraes, C.; Coelho, R.; Gracio, D.; Silva, M.; Peixoto, A.; Lago, P.; Pereira, M.; Catarino, T.; Pinho, S.; et al. Association between Polymorphisms in Antioxidant Genes and Inflammatory Bowel Disease. PLoS ONE 2017, 12, e0169102. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Oshitani, N.; Matsumoto, T.; Nishigami, T.; Arakawa, T.; Inoue, M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br. J. Cancer 2005, 93, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Coelho, R.; Gracio, D.; Dias, C.; Silva, M.; Peixoto, A.; Lopes, P.; Costa, C.; Teixeira, J.P.; Macedo, G.; et al. DNA Damage and Oxidative DNA Damage in Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.Y.; Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Giovannucci, E. Vitamin D and prevention of colorectal adenoma: A meta-analysis. Cancer Epidemiol. Prev. Biomark. 2008, 17, 2958–2969. [Google Scholar] [CrossRef]
- Joo, J.H.; Oh, H.; Kim, M.; An, E.J.; Kim, R.K.; Lee, S.Y.; Kang, D.H.; Kang, S.W.; Keun Park, C.; Kim, H.; et al. NADPH Oxidase 1 Activity and ROS Generation Are Regulated by Grb2/Cbl-Mediated Proteasomal Degradation of NoxO1 in Colon Cancer Cells. Cancer Res. 2016, 76, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Moglia, A.; Schiavo, I.; Donnini, S.; Berta, G.N.; Di Scipio, F.; Perrelli, A.; Fornelli, C.; Trabalzini, L.; Retta, S.F. Yeast-Derived Recombinant Avenanthramides Inhibit Proliferation, Migration and Epithelial Mesenchymal Transition of Colon Cancer Cells. Nutrients 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, H.; Niu, J.; Luo, M.; Gou, Y.; Miao, L.; Zou, Z.; Cheng, Y. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. Int. J. Mol. Sci. 2016, 17, 558. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.D.; Chang, B.; Jin, D.H.; Jung, S.I.; Park, M.Y.; Han, Y.; Yang, Y.; Il Kim, K.; Lim, J.S.; et al. Enhanced antitumor activity of vitamin C via p53 in cancer cells. Free Radic. Biol. Med. 2012, 53, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Rehermann, B. Chronic infections with hepatotropic viruses: Mechanisms of impairment of cellular immune responses. Semin. Liver Dis. 2007, 27, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Dustin, L.B.; Cashman, S.B.; Laidlaw, S.M. Immune control and failure in HCV infection-tipping the balance. J. Leukoc. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; McMahon, B.J. Chronic hepatitis B. Hepatology 2007, 45, 507–539. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Takaki, A.; Tamaki, N.; Maruyama, T.; Onishi, H.; Kobayashi, S.; Nouso, K.; Yasunaka, T.; Koike, K.; Hagihara, H.; et al. Serum oxidative-anti-oxidative stress balance is dysregulated in patients with hepatitis C virus-related hepatocellular carcinoma. Hepatol. Res. 2013, 43, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Squadrito, G.; Cerenzia, G.; Cacciola, I.; Raffa, G.; Craxi, A.; Farinati, F.; Missale, G.; Smedile, A.; Tiribelli, C.; et al. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004, 126, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Urban, S.; Antoni, C.; Bottcher, B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007, 26, 4160–4167. [Google Scholar] [CrossRef]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Doria, M.; Klein, N.; Lucito, R.; Schneider, R.J. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995, 14, 4747–4757. [Google Scholar] [CrossRef]
- Shih, W.L.; Kuo, M.L.; Chuang, S.E.; Cheng, A.L.; Doong, S.L. Hepatitis B virus X protein inhibits transforming growth factor-beta -induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 2000, 275, 25858–25864. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Huh, K.W.; Siddiqui, A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol. Cell. Biol. 2001, 21, 7721–7730. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Kim, Y.J. C-terminal region of HBx is crucial for mitochondrial DNA damage. Cancer Lett. 2013, 331, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Lei, Y.; Chen, H.N.; Chen, Y.; Zhang, T.; Li, K.; Xie, N.; Wang, K.; Feng, X.; Pu, Q.; et al. HBV-induced ROS accumulation promotes hepatocarcinogenesis through Snail-mediated epigenetic silencing of SOCS3. Cell Death Differ. 2016, 23, 616–627. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 2000, 118, 554–559. [Google Scholar] [CrossRef]
- Tsubota, A.; Arase, Y.; Ren, F.; Tanaka, H.; Ikeda, K.; Kumada, H. Genotype may correlate with liver carcinogenesis and tumor characteristics in cirrhotic patients infected with hepatitis B virus subtype adw. J. Med. Virol. 2001, 65, 257–265. [Google Scholar] [CrossRef]
- Muroyama, R.; Kato, N.; Yoshida, H.; Otsuka, M.; Moriyama, M.; Wang, Y.; Shao, R.X.; Dharel, N.; Tanaka, Y.; Ohta, M.; et al. Nucleotide change of codon 38 in the X gene of hepatitis B virus genotype C is associated with an increased risk of hepatocellular carcinoma. J. Hepatol. 2006, 45, 805–812. [Google Scholar] [CrossRef]
- Tatsukawa, M.; Takaki, A.; Shiraha, H.; Koike, K.; Iwasaki, Y.; Kobashi, H.; Fujioka, S.; Sakaguchi, K.; Yamamoto, K. Hepatitis B virus core promoter mutations G1613A and C1653T are significantly associated with hepatocellular carcinoma in genotype C HBV-infected patients. BMC Cancer 2011, 11, 458. [Google Scholar] [CrossRef]
- Pollicino, T.; Cacciola, I.; Saffioti, F.; Raimondo, G. Hepatitis B virus PreS/S gene variants: Pathobiology and clinical implications. J. Hepatol. 2014, 61, 408–417. [Google Scholar] [CrossRef]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Yoshida, H.; Ono-Nita, S.K.; Kato, J.; Goto, T.; Otsuka, M.; Lan, K.; Matsushima, K.; Shiratori, Y.; Omata, M. Activation of intracellular signaling by hepatitis B and C viruses: C-viral core is the most potent signal inducer. Hepatology 2000, 32, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Wu, J. Hepatitis B- and hepatitis C-related hepatocellular carcinomas in the United States: Similarities and differences. Hepat. Mon. 2012, 12, e7635. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic. Biol. Med. 2010, 48, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kobune, M.; Nakamura, T.; Kuroiwa, G.; Takada, K.; Takimoto, R.; Sato, Y.; Fujikawa, K.; Takahashi, M.; Takayama, T.; et al. Normalization of elevated hepatic 8-hydroxy-2′-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001, 61, 8697–8702. [Google Scholar] [PubMed]
- Kato, J.; Miyanishi, K.; Kobune, M.; Nakamura, T.; Takada, K.; Takimoto, R.; Kawano, Y.; Takahashi, S.; Takahashi, M.; Sato, Y.; et al. Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J. Gastroenterol. 2007, 42, 830–836. [Google Scholar] [CrossRef]
- Hino, K.; Nishina, S.; Hara, Y. Iron metabolic disorder in chronic hepatitis C: Mechanisms and relevance to hepatocarcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 4), 93–98. [Google Scholar] [CrossRef]
- Nishina, S.; Hino, K.; Korenaga, M.; Vecchi, C.; Pietrangelo, A.; Mizukami, Y.; Furutani, T.; Sakai, A.; Okuda, M.; Hidaka, I.; et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 2008, 134, 226–238. [Google Scholar] [CrossRef]
- Furutani, T.; Hino, K.; Okuda, M.; Gondo, T.; Nishina, S.; Kitase, A.; Korenaga, M.; Xiao, S.Y.; Weinman, S.A.; Lemon, S.M.; et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 2006, 130, 2087–2098. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Unalp, A.; Behling, C.E.; Lavine, J.E.; Neuschwander-Tetri, B.A. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): A histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009, 49, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Fassio, E.; Alvarez, E.; Dominguez, N.; Landeira, G.; Longo, C. Natural history of nonalcoholic steatohepatitis: A longitudinal study of repeat liver biopsies. Hepatology 2004, 40, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Saibara, T. Clinical features of nonalcoholic steatohepatitis in Japan: Evidence from the literature. J. Gastroenterol. 2006, 41, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.L.; Pagliassotti, M.J. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2008, 19, 567–576. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Sakaguchi, H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. 2005, 33, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D.; Carlson, M. The AMP-activated/SNF1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef] [PubMed]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Ludovica Fracanzani, A.; Gatti, S.; Cairo, G.; Fargion, S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Al-Busafi, S.A.; Bhat, M.; Wong, P.; Ghali, P.; Deschenes, M. Antioxidant therapy in nonalcoholic steatohepatitis. Hepat. Res. Treat. 2012, 2012, 947575. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2014, 15, 7352–7379. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.J.; Angus, P.W.; Yeomans, N.D. Incretin-based therapies for the treatment of non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Nguyen, G.; Park, S.Y.; Le, C.T.; Park, W.S.; Choi, D.H.; Cho, E.H. Metformin ameliorates activation of hepatic stellate cells and hepatic fibrosis by succinate and GPR91 inhibition. Biochem. Biophys. Res. Commun. 2018, 495, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Peyton, K.J.; Shebib, A.R.; Wang, H.; Korthuis, R.J.; Durante, W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H84–H93. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin Gastroenterol Hepatol 2018, 16, 558–566.e552. [Google Scholar] [CrossRef] [PubMed]
- Bubols, G.B.; Vianna Dda, R.; Medina-Remon, A.; von Poser, G.; Lamuela-Raventos, R.M.; Eifler-Lima, V.L.; Garcia, S.C. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar] [PubMed]
- Loguercio, C.; Festi, D. Silybin and the liver: From basic research to clinical practice. World J. Gastroenterol. 2011, 17, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Barbagallo, I.; Godos, J.; Lembo, V.; Currenti, W.; Cina, D.; Avola, R.; D’Orazio, N.; Morisco, F.; Galvano, F.; et al. Silibinin Restores NAD(+) Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver. Nutrients 2017, 9, 86. [Google Scholar] [CrossRef]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949.e8. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Li, J.; Wu, P.W.; Zhou, Y.; Dai, B.; Zhang, P.F.; Zhang, Y.H.; Liu, Y.; Shi, X.L. Rage induces hepatocellular carcinoma proliferation and sorafenib resistance by modulating autophagy. Cell Death Dis. 2018, 9, 225. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Faloppi, L.; De Matteis, S.; Foschi, F.G.; Silvestris, N.; Tovoli, F.; Palmieri, V.; Marisi, G.; Brunetti, O.; Vespasiani-Gentilucci, U.; et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: Validation study and biological rationale. Eur. J. Cancer 2017, 86, 106–114. [Google Scholar] [CrossRef]
- Wan, J.; Liu, T.; Mei, L.; Li, J.; Gong, K.; Yu, C.; Li, W. Synergistic antitumour activity of sorafenib in combination with tetrandrine is mediated by reactive oxygen species (ROS)/Akt signaling. Br. J. Cancer 2013, 109, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, M.; Rossi, R.E.; Mandair, D.; Whyand, T.; Caplin, M.E. Nutrition and pancreatic cancer. Anticancer Res. 2014, 34, 9–21. [Google Scholar] [PubMed]
- Parsi, M.A. Obesity and cholangiocarcinoma. World J. Gastroenterol. WJG 2013, 19, 457–462. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Pairojkul, C.; Pinlaor, S.; Khuntikeo, N.; Wongkham, C.; Sripa, B.; Ma, N.; Vaeteewoottacharn, K.; Furukawa, A.; Kobayashi, H.; et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free Radic. Biol. Med. 2013, 65, 1464–1472. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Ishikawa, H.; Tomono, Y.; Kato, H.; Tsutsumi, K.; Tamaki, N.; Maruyama, T.; Tomofuji, T.; Tsuzaki, R.; et al. Oxidative stress balance is dysregulated and represents an additional target for treating cholangiocarcinoma. Free Radic. Res. 2016, 50, 732–743. [Google Scholar] [CrossRef]
- Peng, P.; Hyder, O.; Firoozmand, A.; Kneuertz, P.; Schulick, R.D.; Huang, D.; Makary, M.; Hirose, K.; Edil, B.; Choti, M.A.; et al. Impact of sarcopenia on outcomes following resection of pancreatic adenocarcinoma. J. Gastrointest. Surg. 2012, 16, 1478–1486. [Google Scholar] [CrossRef]
- Rollins, K.E.; Tewari, N.; Ackner, A.; Awwad, A.; Madhusudan, S.; Macdonald, I.A.; Fearon, K.C.; Lobo, D.N. The impact of sarcopenia and myosteatosis on outcomes of unresectable pancreatic cancer or distal cholangiocarcinoma. Clin. Nutr. 2016, 35, 1103–1109. [Google Scholar] [CrossRef]
- Stretch, C.; Aubin, J.M.; Mickiewicz, B.; Leugner, D.; Al-Manasra, T.; Tobola, E.; Salazar, S.; Sutherland, F.R.; Ball, C.G.; Dixon, E.; et al. Sarcopenia and myosteatosis are accompanied by distinct biological profiles in patients with pancreatic and periampullary adenocarcinomas. PLoS ONE 2018, 13, e0196235. [Google Scholar] [CrossRef]
- Kuang, Y.; Sechi, M.; Nurra, S.; Ljungman, M.; Neamati, N. Design and Synthesis of Novel Reactive Oxygen Species Inducers for the Treatment of Pancreatic Ductal Adenocarcinoma. J. Med. Chem. 2018, 61, 1576–1594. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ignacio Barrasa, J.; Olmo, N.; Perez-Ramos, P.; Santiago-Gomez, A.; Lecona, E.; Turnay, J.; Antonia Lizarbe, M. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis 2011, 16, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, H.; Ahad, A.; Iqbal, J.; Siddiqui, W.A. Pharmacological potential of tocotrienols: A review. Nutr. Metab. 2014, 11, 52. [Google Scholar] [CrossRef]
- Peng, L.; Liu, X.; Lu, Q.; Tang, T.; Yang, Z. Vitamin E intake and pancreatic cancer risk: A meta-analysis of observational studies. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 1249–1255. [Google Scholar] [CrossRef]
- Husain, K.; Centeno, B.A.; Coppola, D.; Trevino, J.; Sebti, S.M.; Malafa, M.P. Delta-Tocotrienol, a natural form of vitamin E, inhibits pancreatic cancer stem-like cells and prevents pancreatic cancer metastasis. Oncotarget 2017, 8, 31554–31567. [Google Scholar] [CrossRef]
- Bast, A.; Haenen, G.R. Ten misconceptions about antioxidants. Trends Pharm. Sci. 2013, 34, 430–436. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed]
- Jakstaite, A.; Maziukiene, A.; Silkuniene, G.; Kmieliute, K.; Dauksa, A.; Paskauskas, S.; Gulbinas, A.; Dambrauskas, Z. Upregulation of cugbp2 increases response of pancreatic cancer cells to chemotherapy. Langenbecks Arch. Surg. 2016, 401, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of Overcoming Intrinsic Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma through the Redox Modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takaki, A.; Kawano, S.; Uchida, D.; Takahara, M.; Hiraoka, S.; Okada, H. Paradoxical Roles of Oxidative Stress Response in the Digestive System before and after Carcinogenesis. Cancers 2019, 11, 213. https://doi.org/10.3390/cancers11020213
Takaki A, Kawano S, Uchida D, Takahara M, Hiraoka S, Okada H. Paradoxical Roles of Oxidative Stress Response in the Digestive System before and after Carcinogenesis. Cancers. 2019; 11(2):213. https://doi.org/10.3390/cancers11020213
Chicago/Turabian StyleTakaki, Akinobu, Seiji Kawano, Daisuke Uchida, Masahiro Takahara, Sakiko Hiraoka, and Hiroyuki Okada. 2019. "Paradoxical Roles of Oxidative Stress Response in the Digestive System before and after Carcinogenesis" Cancers 11, no. 2: 213. https://doi.org/10.3390/cancers11020213
APA StyleTakaki, A., Kawano, S., Uchida, D., Takahara, M., Hiraoka, S., & Okada, H. (2019). Paradoxical Roles of Oxidative Stress Response in the Digestive System before and after Carcinogenesis. Cancers, 11(2), 213. https://doi.org/10.3390/cancers11020213