Mechanistic Modelling of Radiation Responses


Abstract
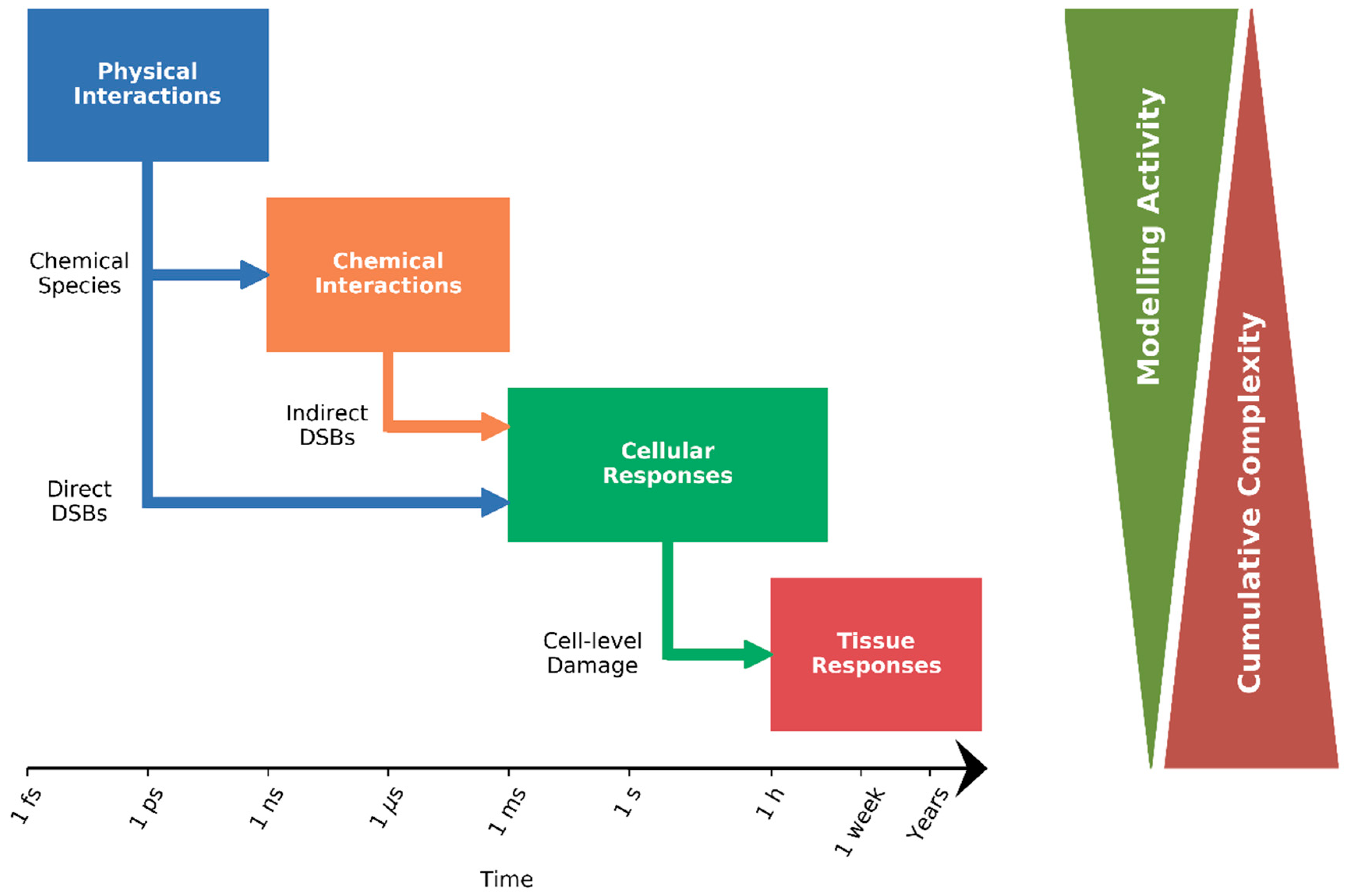
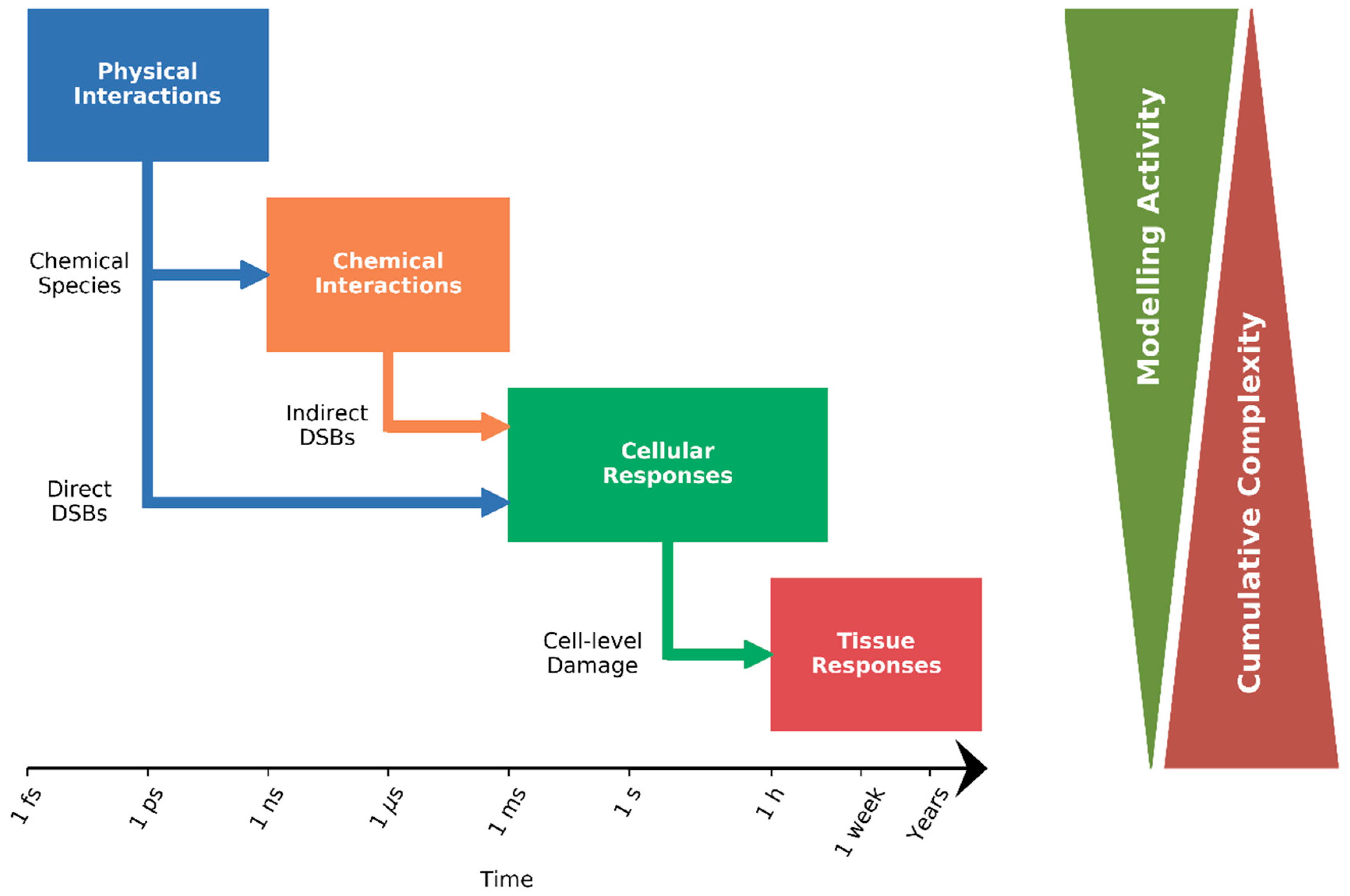
:1. Introduction
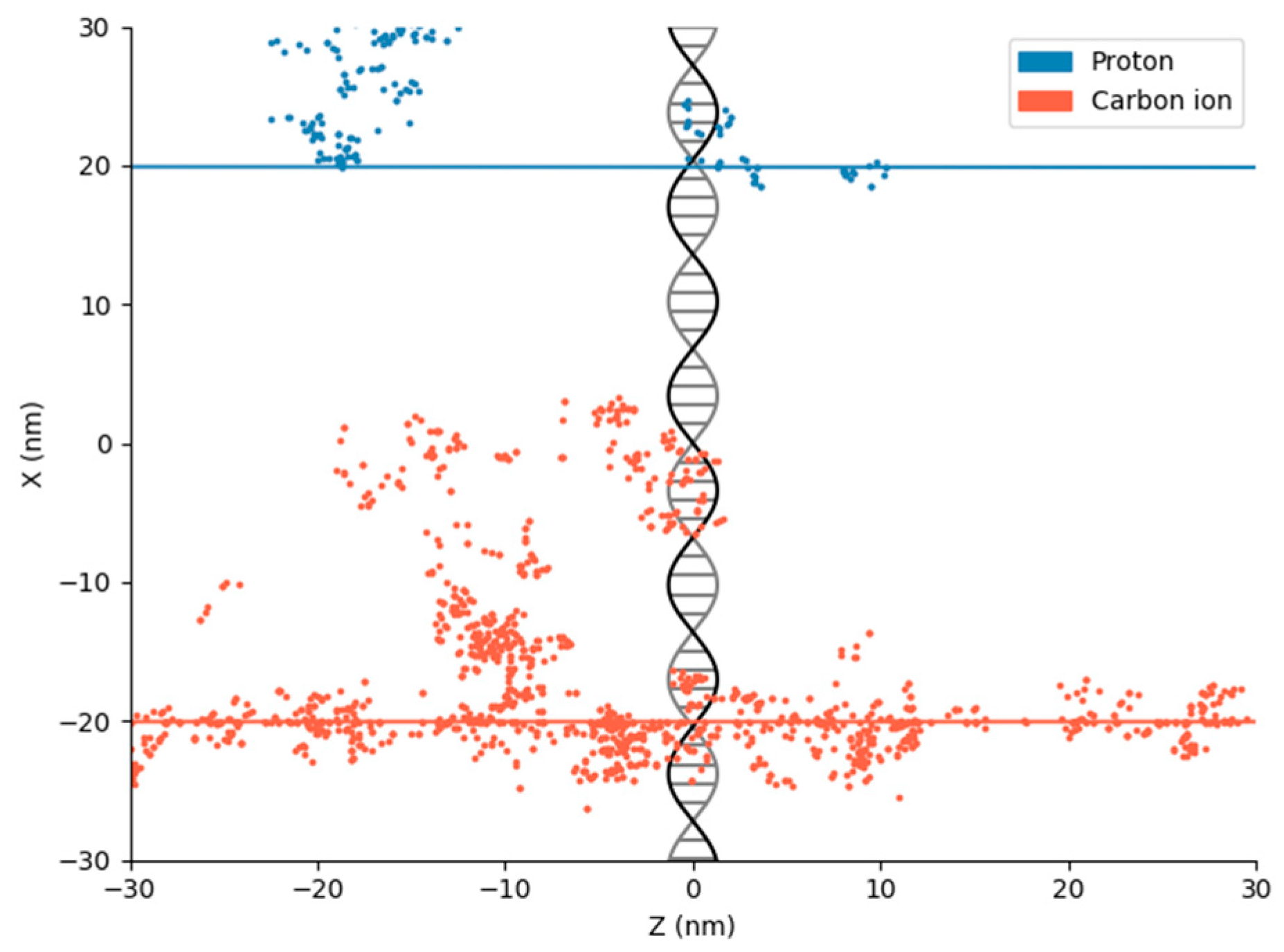
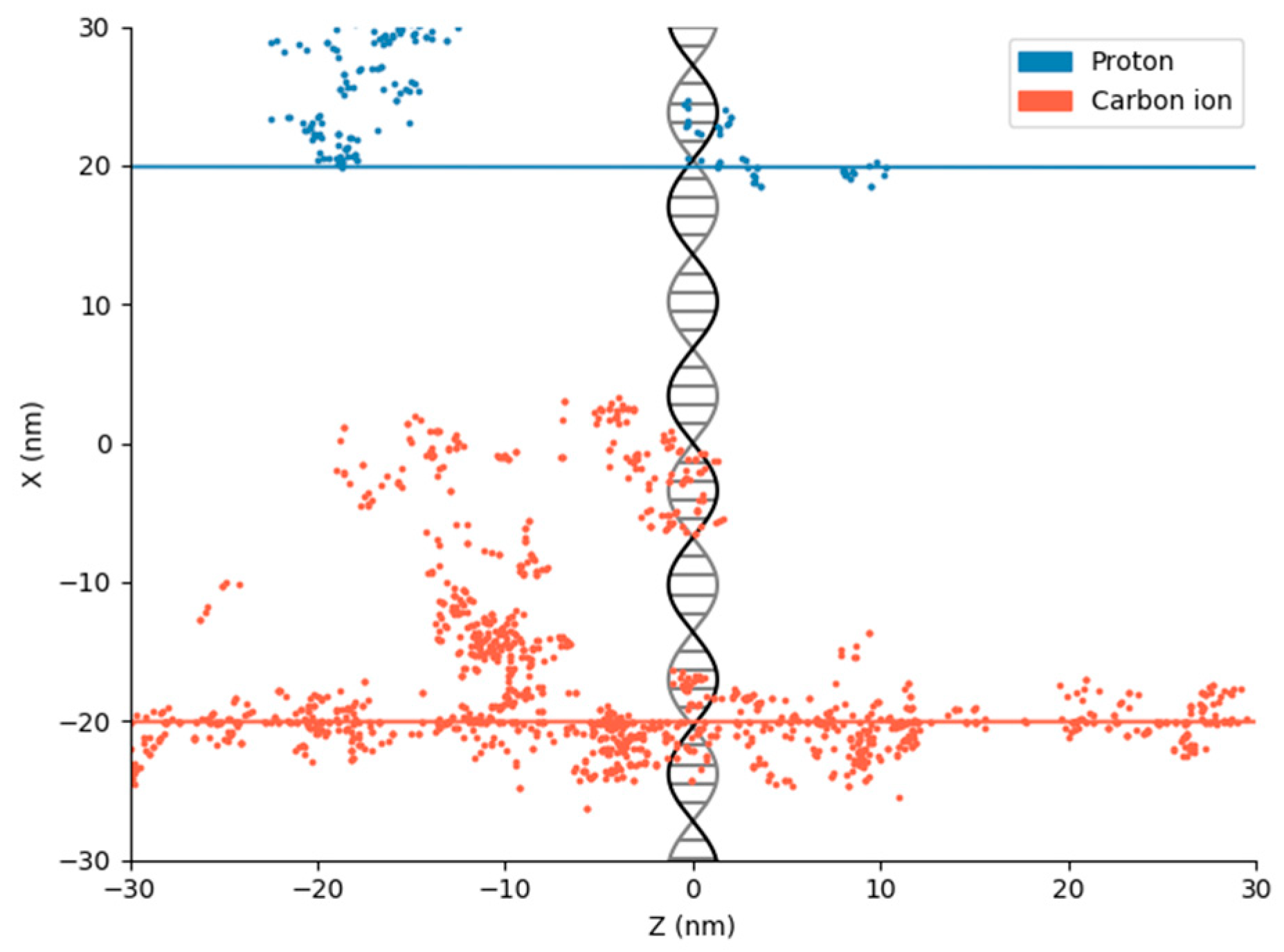
2. Physical DNA Damage
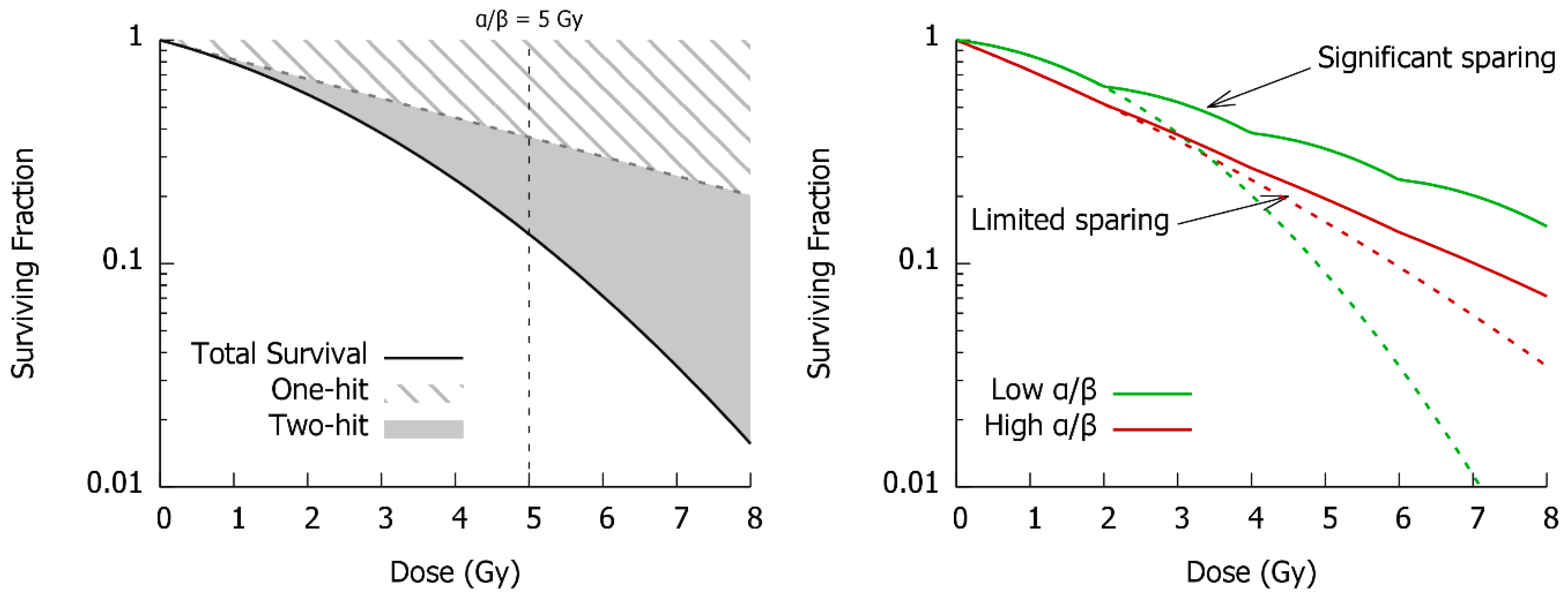
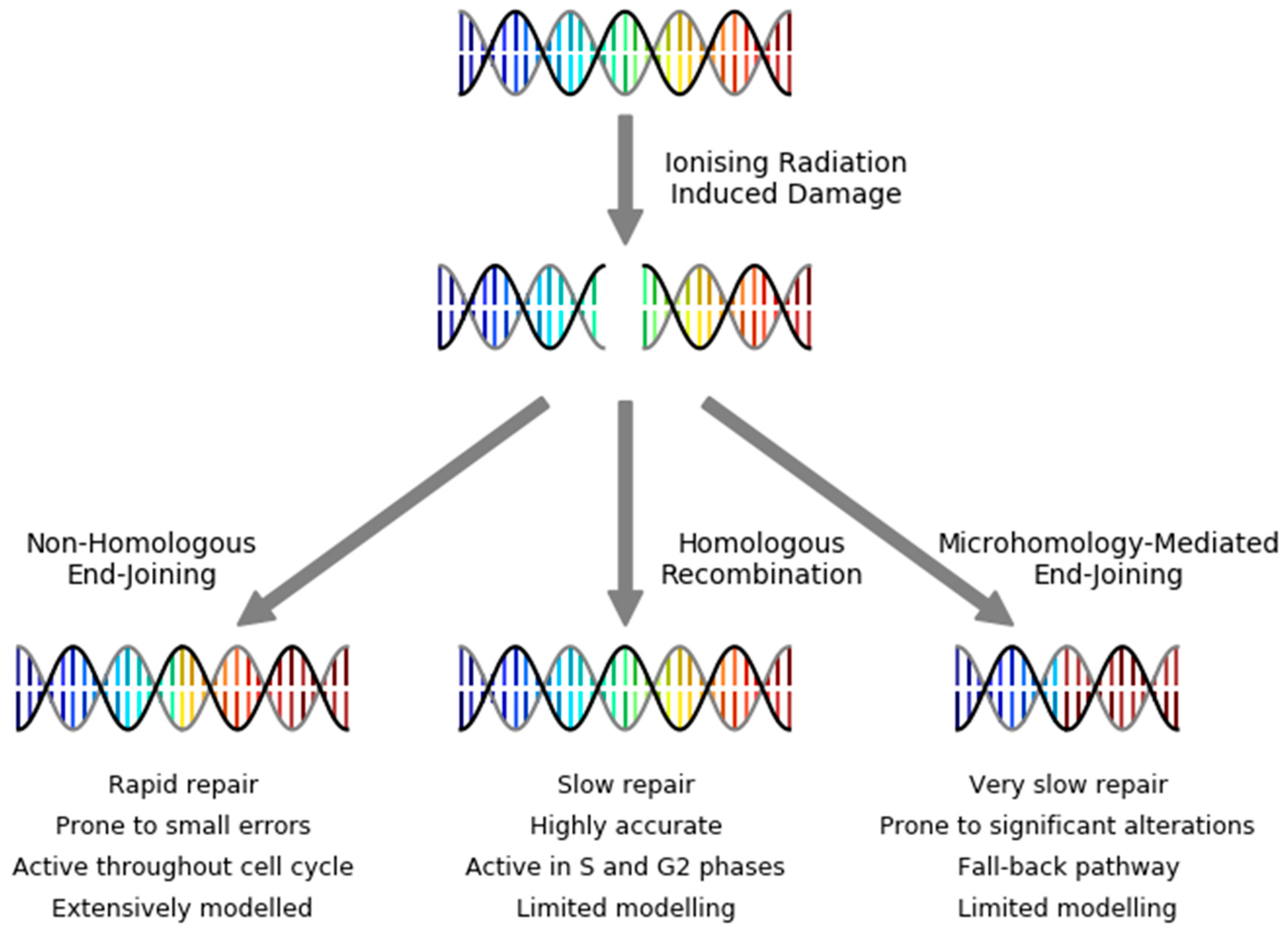
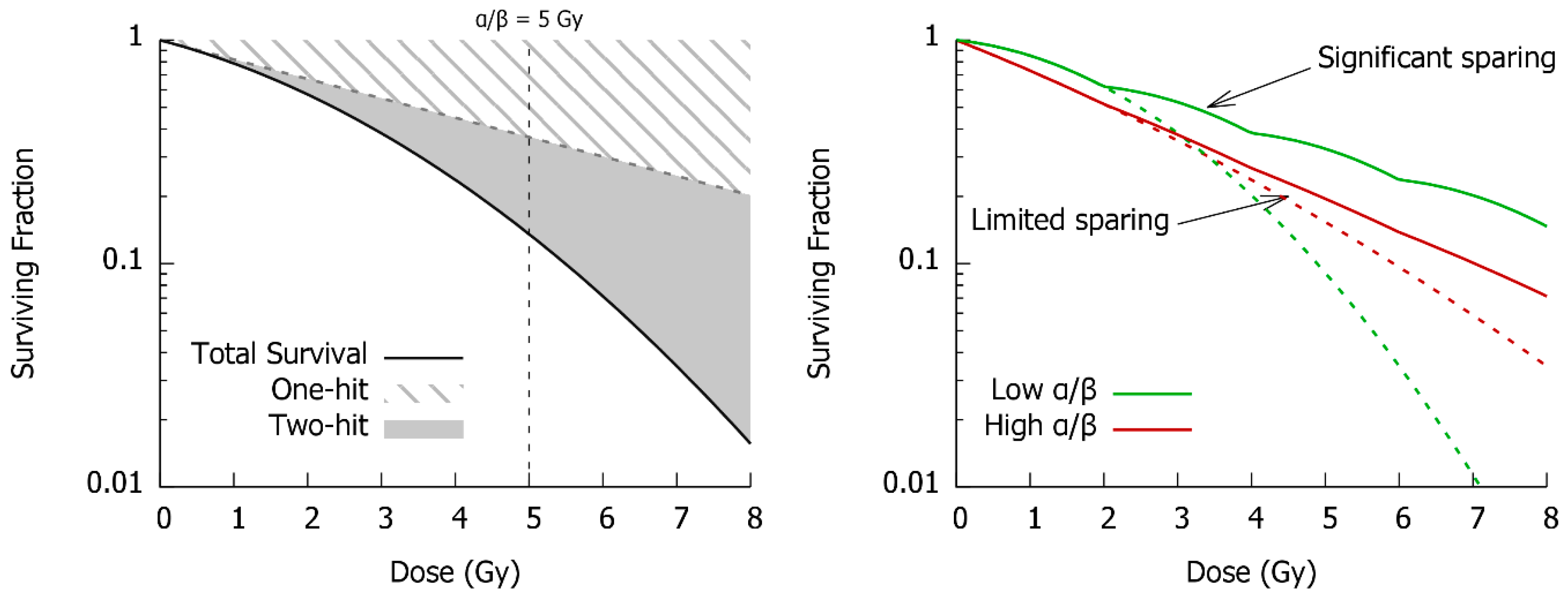
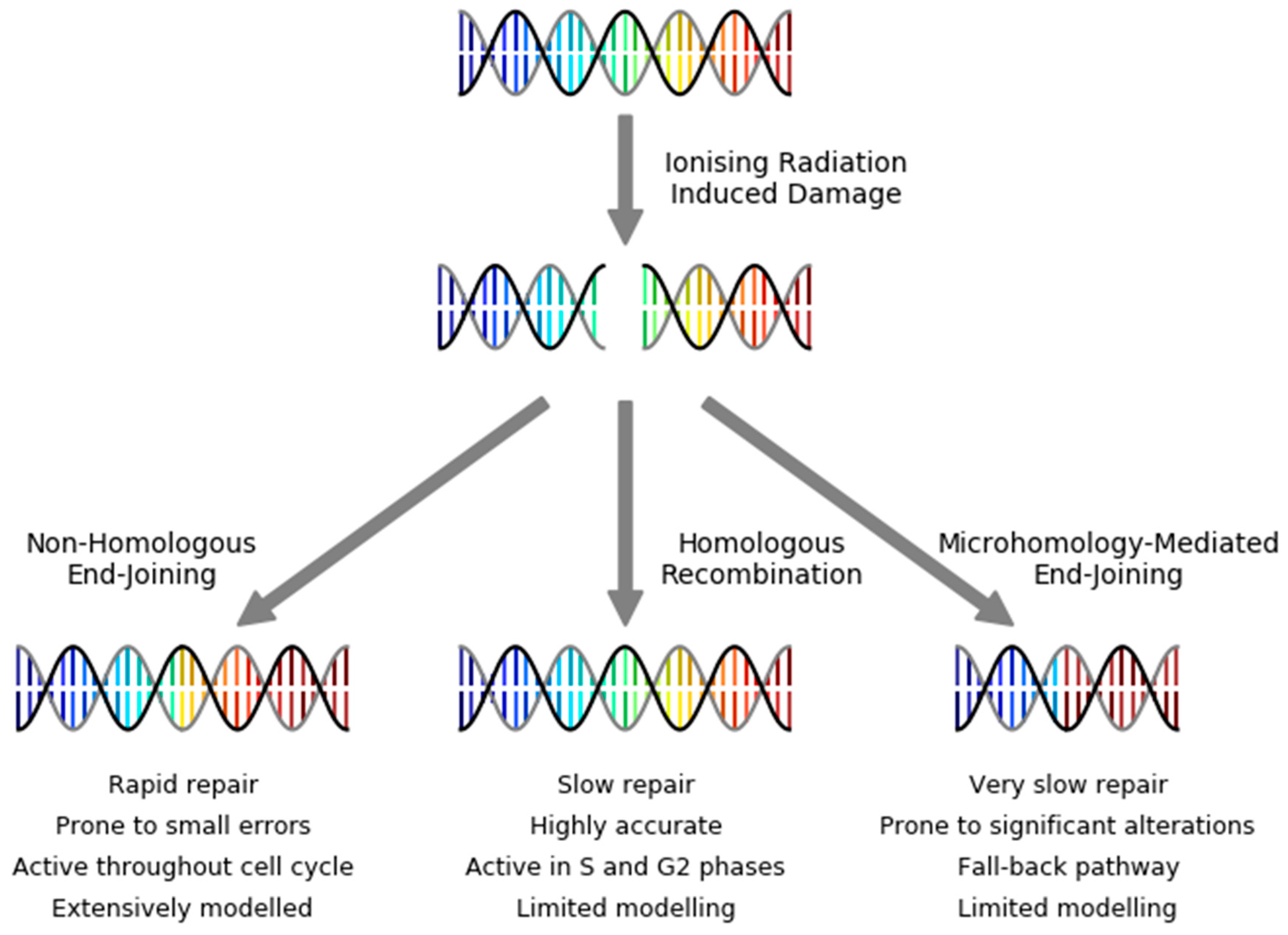
3. DNA Repair
4. Cell Fate
5. Tissue-Level Responses
6. Potential Impacts of Modelling Advances
7. Conclusions
Funding
Conflicts of Interest
References
- Curie, M. Sur l’étude des courbes de probabilité relatives à l’action des rayons X sur les bacilles. Comptes Rendus l’Académie des Sci. 1929, 188, 202–204. (In French) [Google Scholar]
- Crowther, J.A. Some Considerations Relative to the Action of X-Rays on Tissue Cells. Proc. R. Soc. B Biol. Sci. 1924, 96, 207–211. [Google Scholar] [CrossRef]
- Alper, T.; Gillies, N.E.; Elkind, M.M. The sigmoid survival curve in radiobiology. Nature 1960, 186, 1062–1063. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Marcus, P.L. Action of X-rays on Mammalian Cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Morkovin, D.; Marcus, P.I.; Cieciura, S.J. Action of x-rays on mammalian cells. II. Survival curves of cells from normal human tissues. J. Exp. Med. 1957, 106, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Barendsen, G.; Beusker, T.; Vergroesen, A.; Budke, L. Effects of different ionizing radiations on human cells in tissue culture: II. biological experiments. Radiat. Res. 1960, 13, 841–849. [Google Scholar] [CrossRef]
- Sinclair, W.K. The shape of radiation survival curves of mammalian cells cultured in vitro. In Biophysical Aspects of Radiation Quality; International Atomic Energy Agency: Vienna, Austria, 1966. [Google Scholar]
- Douglas, B.G.; Fowler, J.F. The effect of multiple small doses of X rays on skin reactions in the mouse and a basic interpretation. Radiat. Res. 1976, 66, 401–426. [Google Scholar] [CrossRef]
- Barendsen, G.W. Dose fractionation, dose rate and iso-effect relationships for normal tissue responses. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 1981–1997. [Google Scholar] [CrossRef]
- Thames, H.D.; Rodney Withers, H.; Peters, L.J.; Fletcher, G.H. Changes in early and late radiation responses with altered dose fractionation: Implications for dose-survival relationships. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 219–226. [Google Scholar] [CrossRef]
- Fowler, J.F. 21 Years of biologically effective dose. Br. J. Radiol. 2010, 83, 554–568. [Google Scholar] [CrossRef]
- Catcheside, D.G.; Lea, D.E.; Thoday, J.M. The production of chromosome structural changes in Tradescantia microspores in relation to dosage, intensity and temperature. J. Genet. 1946, 47, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Lea, D.E. Actions of Radiations on Living Cells, 1st ed.; Cambridge University Press: London, UK, 1946. [Google Scholar]
- Kellerer, A.M.; Rossi, H.H. The theory of dual radiation action. Curr. Top. Radiat. Res. 1972, 8, 85–158. [Google Scholar]
- Chadwick, K.H.; Leenhouts, H.P. A molecular theory of cell survival. Phys. Med. Biol. 1973, 18, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Tobias, C.A.; Blakelly, E.A.; Ngo, F.Q.H.; Yang, T.C.H. The Repair-Misrepair Model of Cell Survival. In Radiation Biology and Cancer Research; Meyn, A., Withers, R., Eds.; Raven Press: New York, NY, USA, 1980; pp. 195–230. ISBN 978-3-319-46447-3. [Google Scholar]
- Tobias, C.A. The repair-misrepair model in radiobiology: Comparison to other models. Radiat. Res. Suppl. 1985, 8, S77–S95. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.B. Lethal and potentially lethal lesions induced by radiation--a unified repair model. Radiat. Res. 1986, 106, 252–270. [Google Scholar] [CrossRef]
- Goodhead, D.T. Saturable repair models of radiation action in mammalian cells. Radiat. Res. Suppl. 1985, 8, S58–S67. [Google Scholar] [CrossRef]
- Bodgi, L.; Canet, A.; Pujo-Menjouet, L.; Lesne, A.; Victor, J.M.; Foray, N. Mathematical models of radiation action on living cells: From the target theory to the modern approaches. A historical and critical review. J. Theor. Biol. 2016, 394, 93–101. [Google Scholar] [CrossRef]
- Withers, R.H. The Four R’s of Radiotherapy; Academic Press, Inc.: Cambridge, MA, USA, 1975; Volume 5, ISBN 9780120354054. [Google Scholar]
- Loeffler, J.S.; Durante, M. Charged particle therapy—optimization, challenges and future directions. Nat. Rev. Clin. Oncol. 2013, 10, 411–424. [Google Scholar] [CrossRef]
- Sharma, R.A.; Plummer, R.; Stock, J.K.; Greenhalgh, T.A.; Ataman, O.; Kelly, S.; Clay, R.; Adams, R.A.; Baird, R.D.; Billingham, L.; et al. Clinical development of new drug-radiotherapy combinations. Nat. Rev. Clin. Oncol. 2016, 13, 627–642. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Yard, B.D.; Adams, D.J.; Chie, E.E.; Tamayo, P.; Battaglia, J.S.; Gopal, P.; Rogacki, K.; Pearson, B.E.; Phillips, J.; Raymond, D.P.; et al. A genetic basis for the variation in the vulnerability of cancer to DNA Damage. Nat. Commun. 2016, 7, 11428. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial Events in the Cellular Effects of Ionizing-Radiations—Clustered Damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Warters, R.; Hofer, K.; Harris, C.; Smith, J. Radionuclide toxicity in cultured mammalian cells: Elucidation of the primary site of radiation damage. Curr. Top. Radiat. Res. Q. 1978, 12, 389–407. [Google Scholar] [PubMed]
- Munro, T.R. The Relative Radiosensitivity of the Nucleus and Cytoplasm of Chinese Hamster Fibroblasts. Radiat. Res. 1970, 42, 451–470. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F.; Blakely, W.F.; Joner, E.I. Mammalian cells are not killed by DNA single-strand breaks caused by hydroxyl radicals from hydrogen peroxide. Radiat. Res. 1985, 103, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Emfietzoglou, D.; Watanabe, R.; Uehara, S. Can Monte Carlo track structure codes reveal reaction mechanism in DNA damage and improve radiation therapy? Radiat. Phys. Chem. 2008, 77, 1270–1279. [Google Scholar] [CrossRef]
- Zaider, M.; Brenner, D.J.; Wilson, W.E. The applications of track calculations to radiobiology I. Monte Carlo simulation of proton tracks. Radiat. Res. 1983, 95, 231–247. [Google Scholar] [CrossRef]
- Nikjoo, H.; Goodhead, D.T.; Charlton, D.E.; Paretzke, H.G. Energy deposition in small cylindrical targets by ultrasoft X-rays. Phys. Med. Biol. 1989, 34, 691–705. [Google Scholar] [CrossRef]
- Nikjoo, P.; O’Neill, D.T.; Goodhead, H. Computational modelling of low-energy electron-induced DNA damage by early physical and chemical events. Int. J. Radiat. Biol. 1997, 71, 467–483. [Google Scholar] [CrossRef]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational Approach for Determining the Spectrum of DNA Damage Induced by Ionizing Radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Nikjoo, H.; Uehara, S.; Emfietzoglou, D.; Cucinotta, F.A. Track-structure codes in radiation research. Radiat. Meas. 2006, 41, 1052–1074. [Google Scholar] [CrossRef]
- Goorley, T.; James, M.; Booth, T.; Brown, F.; Bull, J.; Cox, L.J.; Durkee, J.; Elson, J.; Fensin, M.; Forster, R.A.; et al. Initial MCNP6 Release Overview. Nucl. Technol. 2012, 180, 298–315. [Google Scholar] [CrossRef]
- Kawrakow, I.; Rogers, D.W.O. The EGSnrc Code System: Monte Carlo Simulation of Electron and Photon Transport; National Research Council Canada: Ottawa, ON, Canada, January 2000. [Google Scholar]
- Battistoni, G.; Bauer, J.; Boehlen, T.T.; Cerutti, F.; Chin, M.P.W.; Dos Santos Augusto, R.; Ferrari, A.; Ortega, P.G.; Kozłowska, W.; Magro, G.; et al. The FLUKA Code: An Accurate Simulation Tool for Particle Therapy. Front. Oncol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Emfietzoglou, D.; Liamsuwan, T.; Taleei, R.; Liljequist, D.; Uehara, S. Radiation track, DNA damage and response—A review. Reports Prog. Phys. 2016, 79. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D.; Papamichael, G.; Nikjoo, H. Monte Carlo Electron Track Structure Calculations in Liquid Water Using a New Model Dielectric Response Function. Radiat. Res. 2017, 188, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Plante, I.; Ponomarev, A.L.; Cucinotta, F.A. Calculation of the energy deposition in nanovolumes by protons and HZE particles: Geometric patterns of initial distributions of DNA repair foci. Phys. Med. Biol. 2013, 58, 6393–6405. [Google Scholar] [CrossRef] [PubMed]
- Wälzlein, C.; Krämer, M.; Scifoni, E.; Durante, M. Low-energy electron transport in non-uniform media. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2014, 320, 75–82. [Google Scholar]
- Sato, T.; Iwamoto, Y.; Hashimoto, S.; Ogawa, T.; Furuta, T.; Abe, S.I.; Kai, T.; Tsai, P.E.; Matsuda, N.; Iwase, H.; et al. Features of Particle and Heavy Ion Transport code System (PHITS) version 3.02. J. Nucl. Sci. Technol. 2018, 55, 684–690. [Google Scholar] [CrossRef]
- Friedland, W.; Dingfelder, M.; Kundrat, P.; Jacob, P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2011, 711, 28–40. [Google Scholar] [CrossRef]
- Agostinelli, S.; Allison, J.; Amako, K.; Apostolakis, J.; Araujo, H.; Arce, P.; Asai, M.; Axen, D.; Banerjee, S.; Barrand, G.; et al. GEANT4—A simulation toolkit. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2003, 506, 250–303. [Google Scholar] [CrossRef]
- Kyriakou, I.; Emfietzoglou, D.; Ivanchenko, V.; Bordage, M.C.; Guatelli, S.; Lazarakis, P.; Tran, H.N.; Incerti, S. Microdosimetry of electrons in liquid water using the low-energy models of Geant4. J. Appl. Phys. 2017, 122. [Google Scholar] [CrossRef]
- Lazarakis, P.; Incerti, S.; Ivanchenko, V.; Kyriakou, I.; Emfietzoglou, D.; Corde, S.; Rosenfeld, A.B.; Lerch, M.; Tehei, M.; Guatelli, S. Investigation of Track Structure and Condensed History physics models for applications in radiation dosimetry on a micro and nano scale in Geant4. Biomed. Phys. Eng. Express 2018, 4, 024001. [Google Scholar] [CrossRef]
- Incerti, S.; Baldacchino, G.; Bernal, M.; Capra, R.; Champion, C.; Francis, Z.; Guèye, P.; Mantero, A.; Mascialino, B.; Moretto, P.; et al. The geant4-dna project. Int. J. Model. Simul. Sci. Comput. 2010, 1, 157–178. [Google Scholar] [CrossRef]
- Bernal, M.A.A.; Bordage, M.C.C.; Brown, J.M.C.M.C.; Davídková, M.; Delage, E.; El Bitar, Z.; Enger, S.A.A.; Francis, Z.; Guatelli, S.; Ivanchenko, V.N.N.; et al. Track structure modeling in liquid water: A review of the Geant4-DNA very low energy extension of the Geant4 Monte Carlo simulation toolkit. Phys. Med. 2015, 31, 1–14. [Google Scholar] [CrossRef]
- Perl, J.; Shin, J.; Schümann, J.; Faddegon, B.; Paganetti, H. TOPAS: An innovative proton Monte Carlo platform for research and clinical applications. Med. Phys. 2012, 39, 6818–6837. [Google Scholar] [CrossRef] [PubMed]
- McNamara, A.; Geng, C.; Turner, R.; Mendez, J.R.; Perl, J.; Held, K.; Faddegon, B.; Paganetti, H.; Schuemann, J. Validation of the radiobiology toolkit TOPAS-nBio in simple DNA geometries. Phys. Med. 2017, 33, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Bug, M.U.; Yong Baek, W.; Rabus, H.; Villagrasa, C.; Meylan, S.; Rosenfeld, A.B. An electron-impact cross section data set (10 eV–1 keV) of DNA constituents based on consistent experimental data: A requisite for Monte Carlo simulations. Radiat. Phys. Chem. 2017, 130, 459–479. [Google Scholar] [CrossRef]
- Francis, Z.; El Bitar, Z.; Incerti, S.; Bernal, M.A.; Karamitros, M.; Tran, H.N. Calculation of lineal energies for water and DNA bases using the Rudd model cross sections integrated within the Geant4-DNA processes. J. Appl. Phys. 2017, 122. [Google Scholar] [CrossRef]
- Karamitros, M.; Luan, S.; Bernal, M.A.; Allison, J.; Baldacchino, G.; Davidkova, M.; Francis, Z.; Friedland, W.; Ivantchenko, V.; Ivantchenko, A.; et al. Diffusion-controlled reactions modeling in Geant4-DNA. J. Comput. Phys. 2014, 274, 841–882. [Google Scholar] [CrossRef]
- Ramos-Méndez, J.; Perl, J.; Schuemann, J.; McNamara, A.; Paganetti, H.; Faddegon, B. Monte Carlo simulation of chemistry following radiolysis with TOPAS-nBio. Phys. Med. Biol. 2018, 63. [Google Scholar] [CrossRef]
- Boscolo, D.; Krämer, M.; Durante, M.; Fuss, M.C.; Scifoni, E. TRAX-CHEM: A pre-chemical and chemical stage extension of the particle track structure code TRAX in water targets. Chem. Phys. Lett. 2018, 698, 11–18. [Google Scholar] [CrossRef]
- McNamara, A.L.; Ramos-Méndez, J.; Perl, J.; Held, K.; Dominguez, N.; Moreno, E.; Henthorn, N.T.; Kirkby, K.J.; Meylan, S.; Villagrasa, C.; et al. Geometrical structures for radiation biology research as implemented in the TOPAS-nBio toolkit. Phys. Med. Biol. 2018, 63. [Google Scholar] [CrossRef]
- Henthorn, N.T.; Warmenhoven, J.W.; Sotiropoulos, M.; Mackay, R.I.; Kirkby, K.J.; Merchant, M.J. Nanodosimetric Simulation of Direct Ion-Induced DNA Damage Using Different Chromatin Geometry Models. Radiat. Res. 2017, 188, 770–783. [Google Scholar] [CrossRef]
- Delage, E.; Pham, Q.T.; Karamitros, M.; Payno, H.; Stepan, V.; Incerti, S.; Maigne, L.; Perrot, Y. PDB4DNA: Implementation of DNA geometry from the Protein Data Bank (PDB) description for Geant4-DNA Monte-Carlo simulations. Comput. Phys. Commun. 2015, 192, 282–288. [Google Scholar] [CrossRef]
- Semenenko, V.A.; Stewart, R.D. A Fast Monte Carlo Algorithm to Simulate the Spectrum of DNA Damages Formed by Ionizing Radiation. Radiat. Res. 2004, 161, 451–457. [Google Scholar] [CrossRef]
- Friedland, W.; Jacob, P.; Bernhardt, P.; Paretzke, H.G.; Dingfelder, M. Simulation of DNA damage after proton irradiation. Radiat Res 2003, 159, 401–410. [Google Scholar] [CrossRef]
- Lampe, N.; Karamitros, M.; Breton, V.; Brown, J.M.C.; Kyriakou, I.; Sakata, D.; Sarramia, D.; Incerti, S. Mechanistic DNA damage simulations in Geant4-DNA part 1: A parameter study in a simplified geometry. Phys. Med. 2018, 48, 135–145. [Google Scholar] [CrossRef]
- Nikjoo, H.; Girard, P. A model of the cell nucleus for DNA damage calculations. Int. J. Radiat. Biol. 2012, 88, 87–97. [Google Scholar] [CrossRef]
- Friedland, W.; Schmitt, E.; Kundrat, P.; Dingfelder, M.; Baiocco, G.; Barbieri, S.; Ottolenghi, A. Comprehensive track-structure based evaluation of DNA damage by light ions from radiotherapy-relevant energies down to stopping. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Bernal, M.A.; Sikansi, D.; Cavalcante, F.; Incerti, S.; Champion, C.; Ivanchenko, V.; Francis, Z. An atomistic geometrical model of the B-DNA configuration for DNA-radiation interaction simulations. Comput. Phys. Commun. 2013, 184, 2840–2847. [Google Scholar] [CrossRef]
- Meylan, S.; Incerti, S.; Karamitros, M.; Tang, N.; Bueno, M.; Clairand, I.; Villagrasa, C. Simulation of early DNA damage after the irradiation of a fibroblast cell nucleus using Geant4-DNA. Sci. Rep. 2017, 7, 11923. [Google Scholar] [CrossRef]
- Prise, K.M.; Folkard, M.; Michael, B.D.; Vojnovic, B.; Brocklehurst, B.; Hopkirk, A.; Munro, I.H. Critical energies for SSB and DSB induction in plasmid DNA by low-energy photons: Action spectra for strand-break induction in plasmid DNA irradiated in vacuum. Int. J. Radiat. Biol. 2000, 76, 881–890. [Google Scholar]
- Schuemann, J.; McNamara, A.L.; Warmenhoven, J.W.; Henthorn, N.T.; Kirkby, K.; Merchant, M.J.; Ingram, S.; Paganetti, H.; Held, K.; Ramos-Mendez, J.; et al. A new Standard DNA Damage (SDD) data format. Radiat. Res. 2019, 191, 76–92. [Google Scholar] [CrossRef]
- Rothkamm, K.; Löbrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; François, M.; Fenech, M.F.; Leifert, W.R. Persistent γH2AX: A promising molecular marker of DNA damage and aging. Mutat. Res.—Rev. Mutat. Res. 2015, 766, 1–19. [Google Scholar] [CrossRef]
- Noda, A.; Hirai, Y.; Hamasaki, K.; Mitani, H.; Nakamura, N.; Kodama, Y. Unrepairable DNA double-strand breaks that are generated by ionising radiation determine the fate of normal human cells. J. Cell Sci. 2012, 125, 5280–5287. [Google Scholar] [CrossRef]
- Cornforth, M.; Bedford, J. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat. Res. 1987, 111, 385–405. [Google Scholar] [CrossRef]
- Chen, A.M.; Lucas, J.N.; Simpson, P.J.; Griffin, C.S.; Savage, J.R.K.; Brenner, D.J.; Hlatky, L.R.; Sachs, R.K. Computer Simulation of Data on Chromosome Aberrations Produced by X Rays or Alpha Particles and Detected by Fluorescence In Situ Hybridization. Radiat. Res. 1997, 148, S93. [Google Scholar] [CrossRef]
- Sachs, R.; Chen, A.; Brenner, D. Review: Proximity effects in the production of chromosome aberrations by ionizing radiation. Int. J. Radiat. Biol. 1997, 71, 1–19. [Google Scholar]
- Tello Cajiao, J.J.; Carante, M.P.; Bernal Rodriguez, M.A.; Ballarini, F. Proximity effects in chromosome aberration induction by low-LET ionizing radiation. DNA Repair 2017, 58, 38–46. [Google Scholar] [CrossRef]
- McMahon, S.J.; Schuemann, J.; Paganetti, H.; Prise, K.M. Mechanistic Modelling of DNA Repair and Cellular Survival Following Radiation-Induced DNA Damage. Sci. Rep. 2016, 6, 33290. [Google Scholar] [CrossRef]
- Edwards, A.A.; Moiseenko, V.V.; Nikjoo, H. On the mechanism of the formation of chromosomal aberrations by ionising radiation. Radiat. Environ. Biophys. 1996, 35, 25–30. [Google Scholar] [CrossRef]
- McMahon, S.J.; McNamara, A.L.; Schuemann, J.; Paganetti, H.; Prise, K.M. A general mechanistic model enables predictions of the biological effectiveness of different qualities of radiation. Sci. Rep. 2017, 7, 10790. [Google Scholar] [CrossRef]
- Ballarini, F.; Carante, M.P. Chromosome aberrations and cell death by ionizing radiation: Evolution of a biophysical model. Radiat. Phys. Chem. 2016, 128, 18–25. [Google Scholar] [CrossRef]
- Waters, C.A.; Strande, N.T.; Wyatt, D.W.; Pryor, J.M.; Ramsden, D.A. Nonhomologous end joining: A good solution for bad ends. DNA Repair 2014, 17, 39–51. [Google Scholar] [CrossRef]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef]
- Löbrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef]
- Iliakis, G. Backup pathways of NHEJ in cells of higher eukaryotes: Cell cycle dependence. Radiother. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef]
- Fowler, J. Is repair of DNA strand break damage from ionizing radiation second-order rather than first-order? A simpler explanation of apparently multiexponential repair. Radiat. Res. 1999, 136, 124–136. [Google Scholar] [CrossRef]
- Foray, N.; Charvet, A.M.; Duchemin, D.; Favaudon, V.; Lavalette, D. The repair rate of radiation-induced DNA damage: A stochastic interpretation based on the Gamma function. J. Theor. Biol. 2005, 236, 448–458. [Google Scholar] [CrossRef]
- Taleei, R.; Girard, P.M.; Sankaranarayanan, K.; Nikjoo, H. The Non-homologous End-Joining (NHEJ) Mathematical Model for the Repair of Double-Strand Breaks: II. Application to Damage Induced by Ultrasoft X Rays and Low-Energy Electrons. Radiat. Res. 2013, 179, 540–548. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Pluth, J.M.; Anderson, J.A.; Harper, J.V; O’Neill, P. Biochemical kinetics model of DSB repair and induction of gamma-H2AX foci by non-homologous end joining. Radiat. Res. 2008, 169, 214–222. [Google Scholar] [CrossRef]
- Carlson, D.J.; Stewart, R.D.; Semenenko, V.A.; Sandison, G.A. Combined Use of Monte Carlo DNA Damage Simulations and Deterministic Repair Models to Examine Putative Mechanisms of Cell Killing. Radiat. Res. 2008, 169, 447–459. [Google Scholar] [CrossRef]
- Friedland, W.; Jacob, P.; Kundrát, P. Stochastic Simulation of DNA Double-Strand Break Repair by Non-homologous End Joining Based on Track Structure Calculations. Radiat. Res. 2010, 173, 677–688. [Google Scholar] [CrossRef]
- Henthorn, N.T.; Warmenhoven, J.W.; Sotiropoulos, M.; Mackay, R.I.; Kirkby, N.F.; Kirkby, K.J.; Merchant, M.J. In Silico Non-Homologous End Joining Following Ion Induced DNA Double Strand Breaks Predicts That Repair Fidelity Depends on Break Density. Sci. Rep. 2018, 8, 2654. [Google Scholar] [CrossRef]
- Taleei, R.; Nikjoo, H. Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat. Res. 2013, 756, 206–212. [Google Scholar] [CrossRef]
- Matos, J.; West, S.C. Holliday junction resolution: Regulation in space and time. DNA Repair 2014, 19, 176–181. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Guerrero, M.; Carlone, M. Mechanistic Formulation of a Lineal-Quadratic-Linear (LQL) Model: Split-Dose Experiments and Exponentially Decaying Sources. Med. Phys. 2009, 36, 2636. [Google Scholar] [CrossRef]
- Zhao, L.; Mi, D.; Hu, B.; Sun, Y. A generalized target theory and its applications. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Bodgi, L.; Foray, N. The nucleo-shuttling of the ATM protein as a basis for a novel theory of radiation response: Resolution of the linear-quadratic model. Int. J. Radiat. Biol. 2016, 92, 117–131. [Google Scholar] [CrossRef]
- Carabe-Fernandez, A.; Dale, R.G.; Jones, B. The incorporation of the concept of minimum RBE (RbEmin) into the linear-quadratic model and the potential for improved radiobiological analysis of high-LET treatments. Int. J. Radiat. Biol. 2007, 83, 27–39. [Google Scholar] [CrossRef]
- Wedenberg, M.; Lind, B.K.; Hårdemark, B. A model for the relative biological effectiveness of protons: The tissue specific parameter α/β of photons is a predictor for the sensitivity to LET changes. Acta Oncol. 2013, 52, 580–588. [Google Scholar] [CrossRef]
- McNamara, A.L.; Schuemann, J.; Paganetti, H. A phenomenological relative biological effectiveness (RBE) model for proton therapy based on all published in vitro cell survival data. Phys. Med. Biol. 2015, 60, 8399–8416. [Google Scholar] [CrossRef]
- Butts, J.J.; Katz, R. Theory of RBE for Heavy Ion Bombardment of Dry Enzymes and Viruses. Radiat. Res. 1967, 30, 855. [Google Scholar] [CrossRef]
- Elsässer, T.; Krämer, M.; Scholz, M. Accuracy of the local effect model for the prediction of biologic effects of carbon ion beams in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 866–872. [Google Scholar] [CrossRef]
- Friedrich, T.; Scholz, U.; Elssser, T.; Durante, M.; Scholz, M. Calculation of the biological effects of ion beams based on the microscopic spatial damage distribution pattern. Int. J. Radiat. Biol. 2012, 88, 103–107. [Google Scholar] [CrossRef]
- Hawkins, R.B. A microdosimetric-kinetic model of cell death from exposure to ionizing radiation of any LET, with experimental and clinical applications. Int. J. Radiat. Biol. 1996, 69, 739–755. [Google Scholar] [CrossRef]
- Krämer, M.; Scholz, M. Treatment planning for heavy-ion radiotherapy: Calculation and optimization of biologically effective dose. Phys. Med. Biol. 2000, 45, 3319–3330. [Google Scholar] [CrossRef]
- Inaniwa, T.; Furukawa, T.; Kase, Y.; Matsufuji, N.; Toshito, T.; Matsumoto, Y.; Furusawa, Y.; Noda, K. Treatment planning for a scanned carbon beam with a modified microdosimetric kinetic model. Phys. Med. Biol. 2010, 55, 6721–6737. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.B.; Inaniwa, T. A Microdosimetric-Kinetic Model for Cell Killing by Protracted Continuous Irradiation II: Brachytherapy and Biologic Effective Dose. Radiat. Res. 2014, 182, 72–82. [Google Scholar] [CrossRef]
- Sato, T.; Masunaga, S.I.; Kumada, H.; Hamada, N. Microdosimetric Modeling of Biological Effectiveness for Boron Neutron Capture Therapy Considering Intra- and Intercellular Heterogeneity in10B Distribution. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef]
- McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Coulter, J.A.; Jain, S.; Butterworth, K.T.; Schettino, G.; Dickson, G.R.; Hounsell, A.R.; O’Sullivan, J.M.; et al. Biological consequences of nanoscale energy deposition near irradiated heavy atom nanoparticles. Sci. Rep. 2011, 1, 18. [Google Scholar] [CrossRef]
- Friedrich, T.; Durante, M.; Scholz, M. Modeling Cell Survival after Photon Irradiation Based on Double-Strand Break Clustering in Megabase Pair Chromatin Loops. Radiat. Res. 2012, 178, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Verkhovtsev, A.; Surdutovich, E.; Solov’yov, A.V. Multiscale approach predictions for biological outcomes in ion-beam cancer therapy. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Frese, M.C.; Yu, V.K.; Stewart, R.D.; Carlson, D.J. A Mechanism-Based Approach to Predict the Relative Biological Effectiveness of Protons and Carbon Ions in Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2011, 83, 442–450. [Google Scholar] [CrossRef]
- Olive, P.L. Retention of γH2AX foci as an indication of lethal DNA damage. Radiother. Oncol. 2011, 101, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Banáth, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual γH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef]
- Cohen–Jonathan, E.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999, 3, 77–83. [Google Scholar] [CrossRef]
- Watters, D. Molecular mechanisms of ionizing radiation-induced apoptosis. Immunol. Cell Biol. 1999, 77, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, M.; Bhatt, A.N.; Das, A.; Dwarakanath, B.S.; Sharma, K. Radiation-induced autophagy: Mechanisms and consequences. Free Radic. Res. 2016, 50, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Radford, I.R. Mouse lymphoma cells that undergo interphase death show markedly increased sensitivity to radiation-induced DNA double-Strand breakage as compared with cells that undergo mitotic death. Int. J. Radiat. Biol. 1991, 59, 1353–1369. [Google Scholar] [CrossRef]
- Langley, R.E.; Bump, E.A.; Quartuccio, S.G.; Medeiros, D.; Braunhut, S.J. Radiation-induced apoptosis in microvascular endothelial cells. Br. J. Cancer 1997, 75, 666–672. [Google Scholar] [CrossRef]
- Lindner, A.U.; Concannon, C.G.; Boukes, G.J.; Cannon, M.D.; Llambi, F.; Ryan, D.; Boland, K.; Kehoe, J.; McNamara, D.A.; Murray, F.; et al. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013, 73, 519–528. [Google Scholar] [CrossRef]
- Liu, C.; Li, C.-Y.; Yuan, F. Mathematical modeling of the phoenix rising pathway. PLoS Comput. Biol. 2014, 10, e1003461. [Google Scholar] [CrossRef]
- Dolan, D.W.P.; Zupanic, A.; Nelson, G.; Hall, P.; Miwa, S.; Kirkwood, T.B.L.; Shanley, D.P. Integrated Stochastic Model of DNA Damage Repair by Non-homologous End Joining and p53/p21- Mediated Early Senescence Signalling. PLoS Comput. Biol. 2015, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lou, I.C.; Conolly, R.B. Computational modeling of signaling pathways mediating cell cycle checkpoint control and apoptotic responses to ionizing radiation-induced DNA damage. Dose-Response 2012, 10, 251–273. [Google Scholar] [CrossRef] [PubMed]
- Würstle, M.L.; Zink, E.; Prehn, J.H.M.; Rehm, M. From computational modelling of the intrinsic apoptosis pathway to a systems-based analysis of chemotherapy resistance: Achievements, perspectives and challenges in systems medicine. Cell Death Dis. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.; Davidson, S.E.; Roberts, S.A.; Hunter, R.D. The independence of intrinsic radiosensitivity as a prognostic factor for patient response to radiotherapy of carcinoma of the cervix. Br. J. Cancer 1997, 76, 1184–1190. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Zaidi, S.T.; Zietman, A. Multivariate determinants of radiocurability 1: Prediction of single fraction tumor control doses. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 57–66. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Vijayappa, S.; Kurimasa, A.; Ogawa, K.; Chen, D.J. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006, 66, 8352–8355. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.; Nahum, A.E. A model for calculating tumour control probability in radiotherapy including the effects of inhomogeneous distributions of dose and clonogenic cell density. Phys. Med. Biol. 1993, 38, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Gay, H.A.; Niemierko, A. A free program for calculating EUD-based NTCP and TCP in external beam radiotherapy. Phys. Med. 2007, 23, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Niemierko, A. Reporting and analyzing dose distributions: A concept of equivalent uniform dose. Med. Phys. 1997, 24, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Lyman, J.T. Complication probability as assessed from dose-volume histograms. Radiat. Res. Suppl. 1985, 8, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Kutcher, G.J.; Emami, B.; Goitein, M. Fitting of normal tissue tolerance data to an analytic function. Int. J. Radiat. Oncol. Biol. Phys. 1991, 21, 123–135. [Google Scholar] [CrossRef]
- Källman, P.; Ågren, A.; Brahme, A. Tumour and normal tissue responses to fractionated non-uniform dose delivery. Int. J. Radiat. Biol. 1992, 62, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Zaider, M.; Zelefsky, M.J.; Hanin, G.; Li, X.A.; Wang, J.Z.; Stewart, R.D. Tumour control probability: A formulation applicable to any temporal protocol of dose delivery. Phys. Med. Biol. 2000, 45, 279. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Shoghi, K.I.; Deasy, J.O. Modelling the interplay between hypoxia and proliferation in radiotherapy tumour response. Phys. Med. Biol. 2013, 58, 4897–4919. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, E.; Baker, C.; Nahum, A. Mechanistic simulation of normal-tissue damage in radiotherapy—Implications for dose-volume analyses. Phys. Med. Biol. 2010, 55, 2121–2136. [Google Scholar] [CrossRef] [PubMed]
- Little, M.P.; Heidenreich, W.F.; Moolgavkar, S.H.; Schöllnberger, H.; Thomas, D.C. Systems biological and mechanistic modelling of radiation-induced cancer. Radiat. Environ. Biophys. 2008, 47, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Shuryak, I.; Brenner, D.J.; Ullrich, R.L. Radiation-Induced carcinogenesis: Mechanistically based differences between Gamma-Rays and neutrons, and interactions with DMBA. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Rühm, W.; Eidemüller, M.; Kaiser, J. Biologically-based mechanistic models of radiation-related carcinogenesis applied to epidemiological data. Int. J. Radiat. Biol. 2017, 93, 1093–1117. [Google Scholar] [CrossRef]
- Schneider, U. Mechanistic model of radiation-induced cancer after fractionated radiotherapy using the linear-quadratic formula. Med. Phys. 2009, 36, 1138–1143. [Google Scholar] [CrossRef]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Ebert, M.; Suchowerska, N.; Jackson, M.; McKenzie, D.R. A mathematical framework for separating the direct and bystander components of cellular radiation response. Acta Oncol. 2010, 49, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.J.; Butterworth, K.T.; McGarry, C.K.; Trainor, C.; O’Sullivan, J.M.; Hounsell, A.R.; Prise, K.M. A Computational Model of Cellular Response to Modulated Radiation Fields. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 250–256. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.J.; Butterworth, K.T.; Trainor, C.; McGarry, C.K.; O’Sullivan, J.M.; Schettino, G.; Hounsell, A.R.; Prise, K.M. A Kinetic-Based Model of Radiation-Induced Intercellular Signalling. PLoS ONE 2013, 8, e54526. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Formenti, S.C. Radiation as an immunological adjuvant: Current evidence on dose and fractionation. Front. Oncol. 2012, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.J.; Minn, A.J.; Vonderheide, R.H.; Wherry, E.J.; Hahn, S.M.; Maity, A. Awakening the immune system with radiation: Optimal dose and fractionation. Cancer Lett. 2015, 368, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Serre, R.; Benzekry, S.; Padovani, L.; Meille, C.; Andre, N.; Ciccolini, J.; Barlesi, F.; Muracciole, X.; Barbolosi, D. Mathematical modeling of cancer immunotherapy and its synergy with radiotherapy. Cancer Res. 2016, 76, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Comparison between in vitro radiosensitivity and in vivo radioresponse in murine tumor cell lines II: In vivo radioresponse following fractionated treatment and in vitro/in vivo correlations. Int. J. Radiat. Oncol. Biol. Phys. 1990, 18, 331–345. [Google Scholar] [CrossRef]
- West, C.M.; Davidson, S.E.; Roberts, S.A.; Hunter, R.D. Intrinsic radiosensitivity and prediction of patient response to radiotherapy for carcinoma of the cervix. Br. J. Cancer 1993, 68, 819–823. [Google Scholar] [CrossRef]
- Fertil, B.; Malaise, E.P. Inherent cellular radiosensitivity as a basic concept for human tumor radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 621–629. [Google Scholar] [CrossRef]
- Kelland, L.R.; Tonkin, K.S.; Steel, G.G. A comparison of the in vivo and in vitro radiation response of three human cervix carcinomas. Radiother. Oncol. 1989, 16, 55–63. [Google Scholar] [CrossRef]
- Torres-Roca, J.F.; Stevens, C.W. Predicting Response to Clinical Radiotherapy: Past, Present, and Future Directions. Cancer Control 2008, 15, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G.; Berglund, A.; Schell, M.J.; Mihaylov, I.; Fulp, W.J.; Yue, B.; Welsh, E.; Caudell, J.J.; Ahmed, K.; Strom, T.S.; et al. A genome-based model for adjusting radiotherapy dose (GARD): A retrospective, cohort-based study. Lancet Oncol. 2017, 2045, 202–211. [Google Scholar] [CrossRef]
- Torres-Roca, J.F.; Eschrich, S.; Zhao, H.; Bloom, G.; Sung, J.; McCarthy, S.; Cantor, A.B.; Scuto, A.; Li, C.; Zhang, S.; et al. Prediction of Radiation Sensitivity Using a Gene Expression Classifier. Cancer Res. 2005, 65, 7169–7176. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.S.; Iype, R.; Senra, J.; Taylor, J.; Armenoult, L.; Oguejiofor, K.; Li, Y.; Stratford, I.; Stern, P.L.; O’Connor, M.J.; et al. Investigation of Radiosensitivity Gene Signatures in Cancer Cell Lines. PLoS ONE 2014, 9, e86329. [Google Scholar] [CrossRef] [PubMed]
- Forker, L.J.; Choudhury, A.; Kiltie, A.E. Biomarkers of Tumour Radiosensitivity and Predicting Benefit from Radiotherapy. Clin. Oncol. 2015, 27, 561–569. [Google Scholar] [CrossRef]
- Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef]
- Dungey, F.A.; Caldecott, K.W.; Chalmers, A.J. Enhanced radiosensitisation of human glioma cells by combining inhibition of PARP with inhibition of Hsp90. Mol. Cancer Ther. 2010, 8, 2243–2254. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.M.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef]
- van der Schaaf, A.; Langendijk, J.A.; Fiorino, C.; Rancati, T. Embracing Phenomenological Approaches to Normal Tissue Complication Probability Modeling: A Question of Method. Int. J. Radiat. Oncol. 2015, 91, 468–471. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paper | Physics Code | DNA Model | Endpoints |
|---|---|---|---|
| Nikjoo et al. [39,63] | KURBUC/PITS | Whole nucleus containing chromatin fibers arranged in hierarchy of spherical volumes | DSBs, SSBs, base damages |
| Friedland et al. [44,64] | PARTRAC | Whole nucleus containing model chromatin fiber random walk | DSBs, SSBs, DNA fragment sizes |
| Bernal et al. [65] | Geant4-DNA | Atomistic DNA segment model in cube | DSBs, SSBs |
| Plante et al. [41] | RITRACKS | Flexible polymer chain chromosome model in nucleus | DSBs |
| Meylan et al. [66] | Geant4-DNA | DNAFabric-based nucleus model | DSBs, SSBs |
| McNamara et al. [51,57] | Geant4-DNA/TOPAS-nBio | Multiple DNA structures, fractal walk nucleus | DSBs, SSBs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMahon, S.J.; Prise, K.M. Mechanistic Modelling of Radiation Responses. Cancers 2019, 11, 205. https://doi.org/10.3390/cancers11020205
McMahon SJ, Prise KM. Mechanistic Modelling of Radiation Responses. Cancers. 2019; 11(2):205. https://doi.org/10.3390/cancers11020205
Chicago/Turabian StyleMcMahon, Stephen J., and Kevin M. Prise. 2019. "Mechanistic Modelling of Radiation Responses" Cancers 11, no. 2: 205. https://doi.org/10.3390/cancers11020205
APA StyleMcMahon, S. J., & Prise, K. M. (2019). Mechanistic Modelling of Radiation Responses. Cancers, 11(2), 205. https://doi.org/10.3390/cancers11020205