SYK Inhibition Potentiates the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells In Vitro

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results
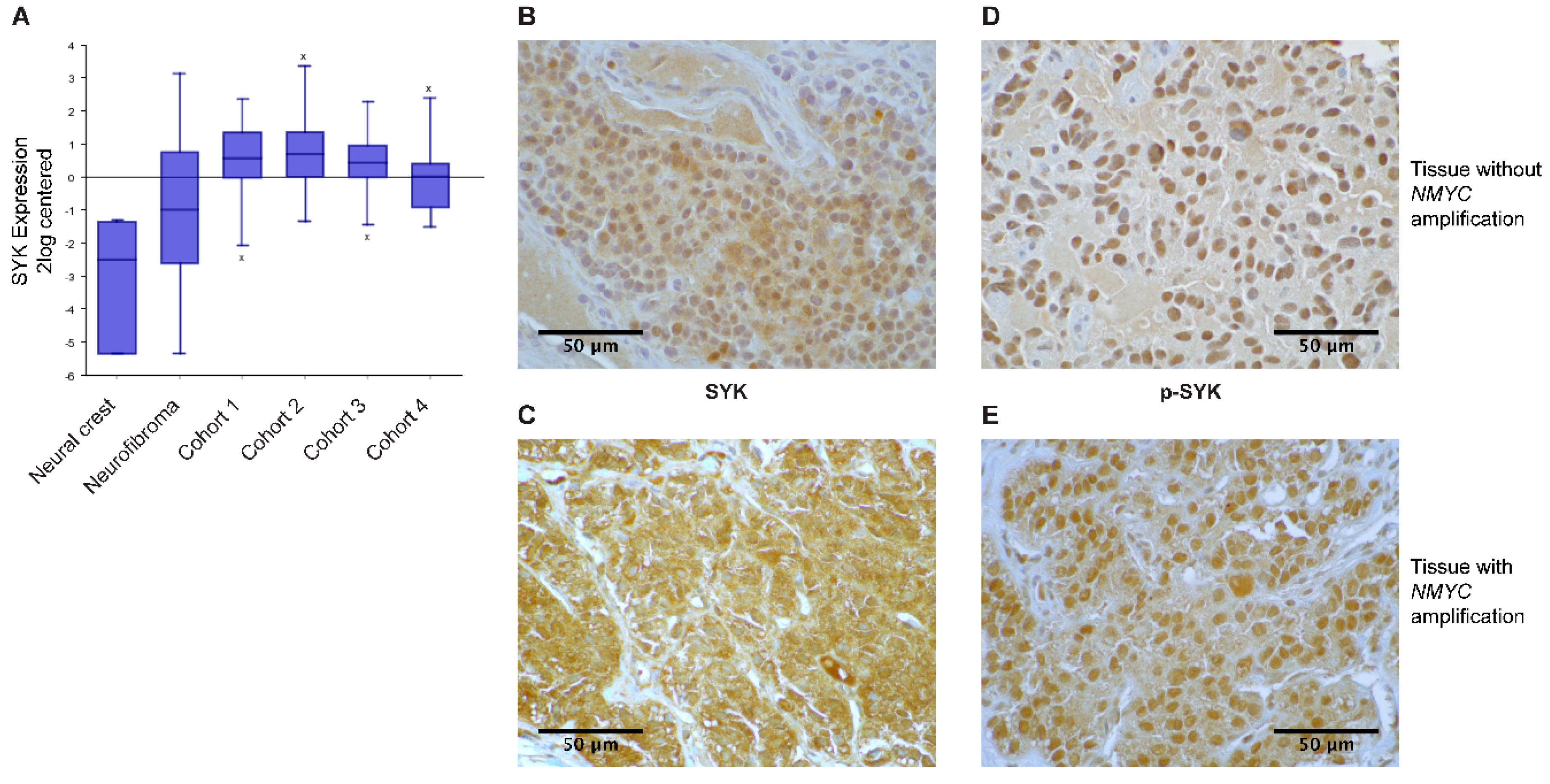
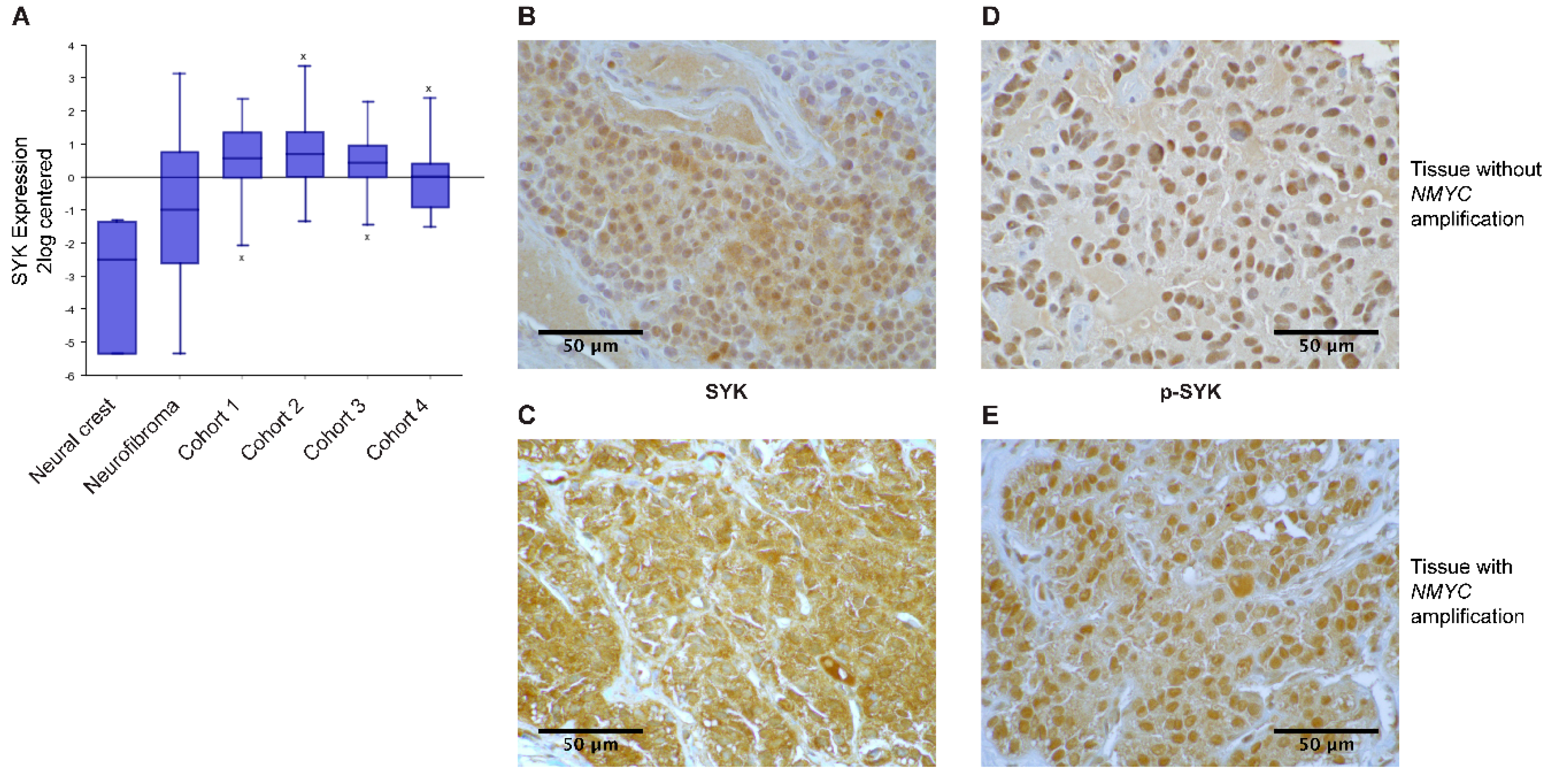
2.1. SYK Is Expressed in Neuroblastoma Tissue
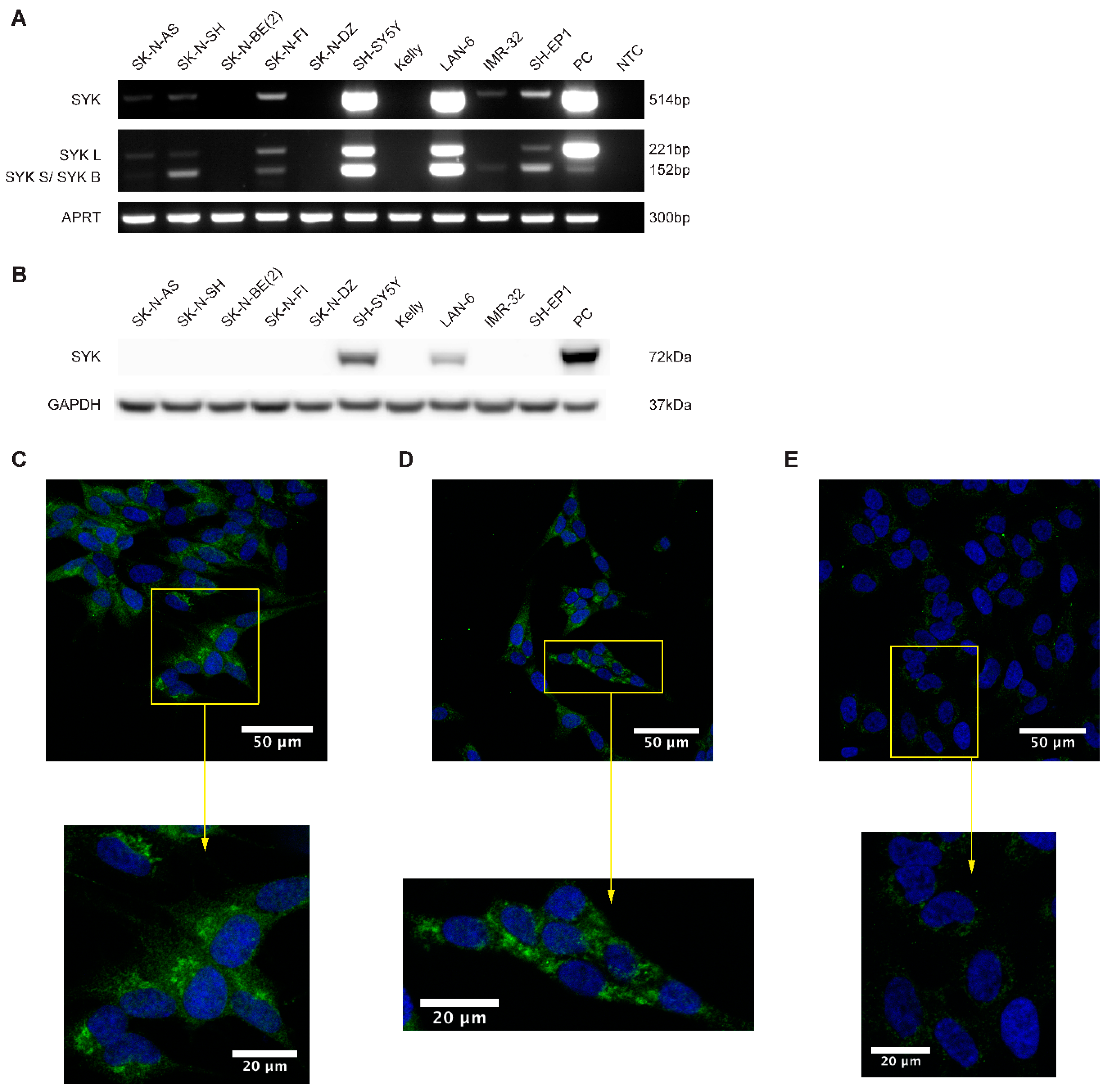
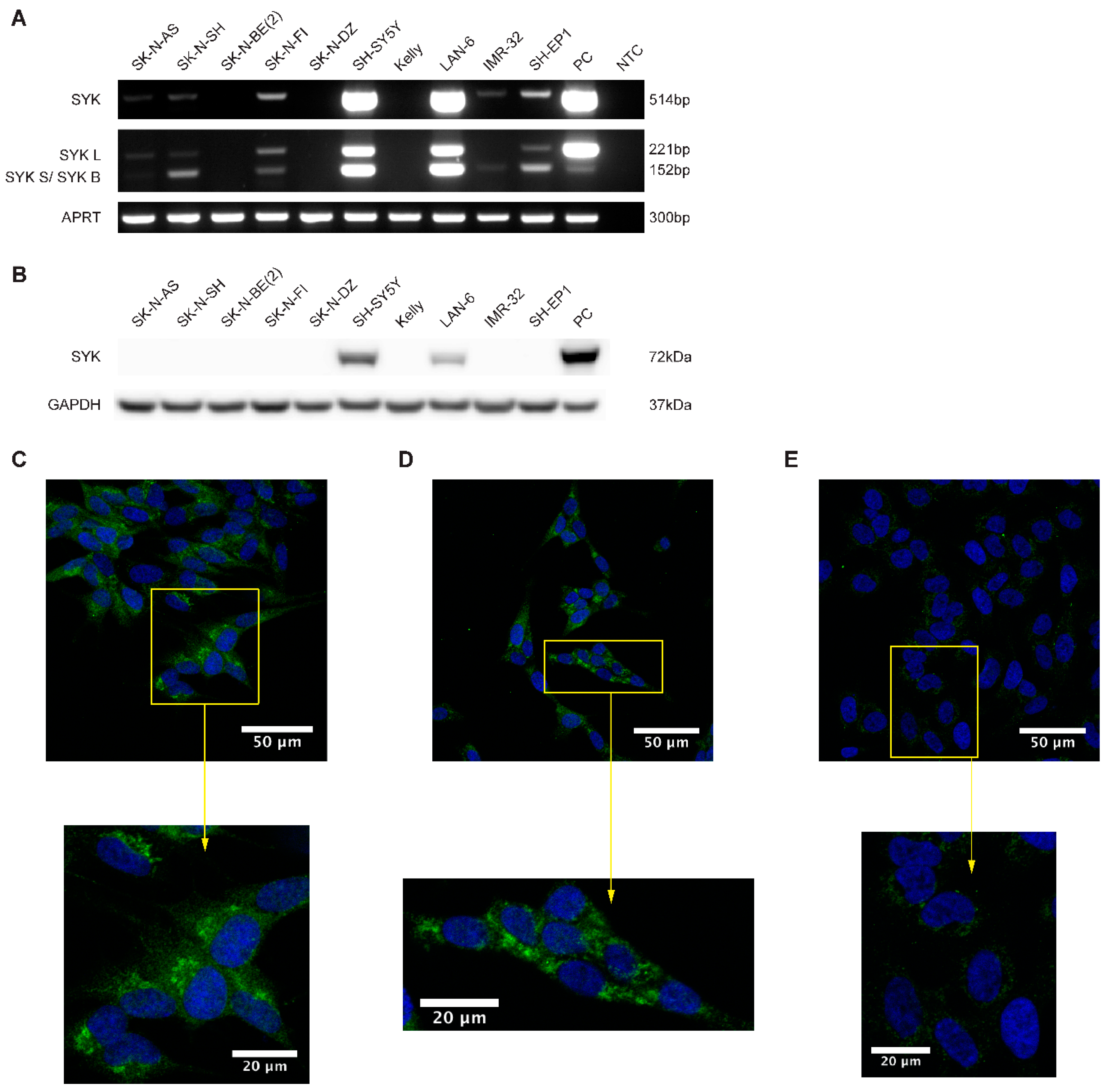
2.2. SYK mRNA and to a Lesser Extend SYK Protein Are Present in Neuroblastoma Cell Lines
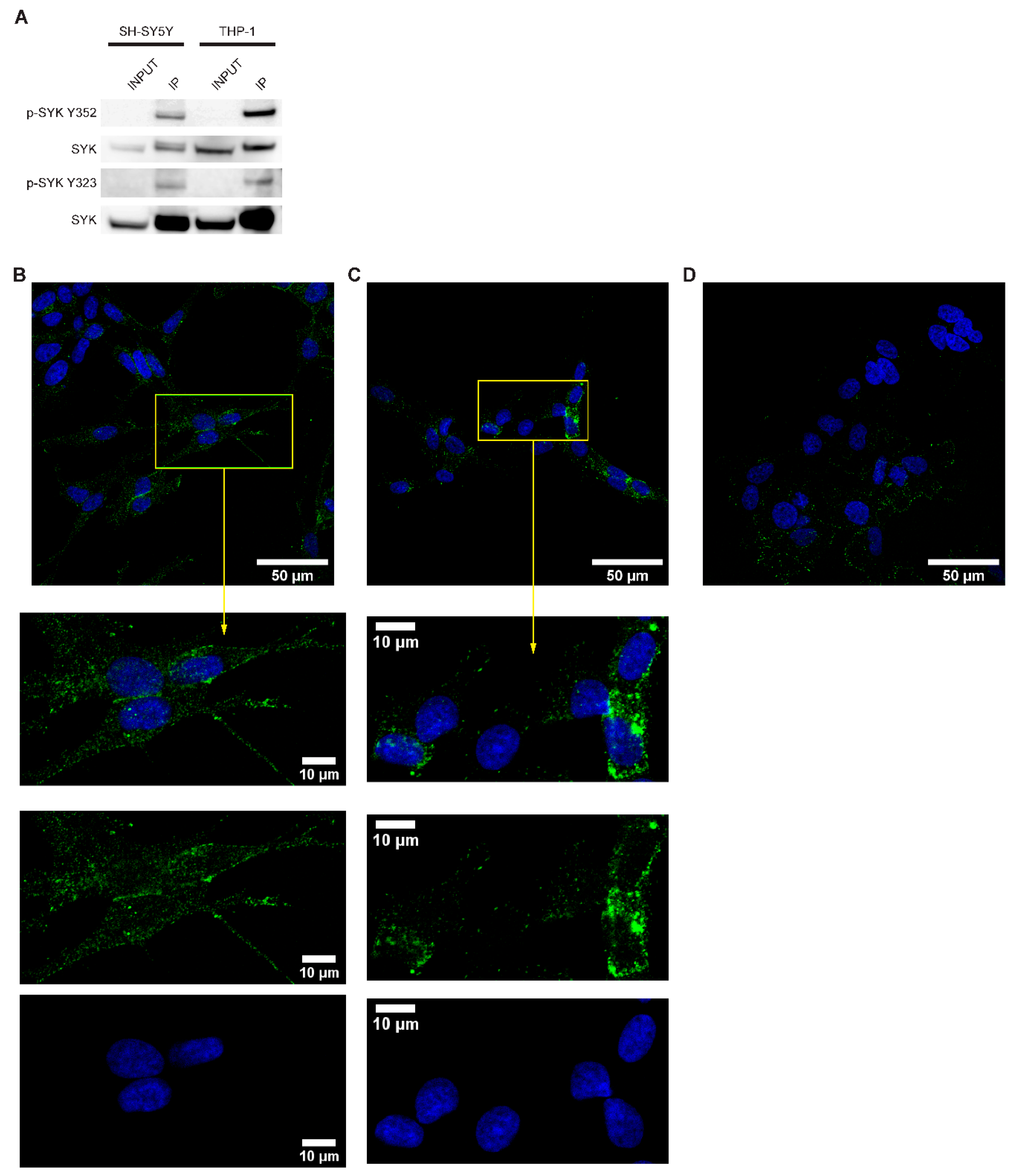
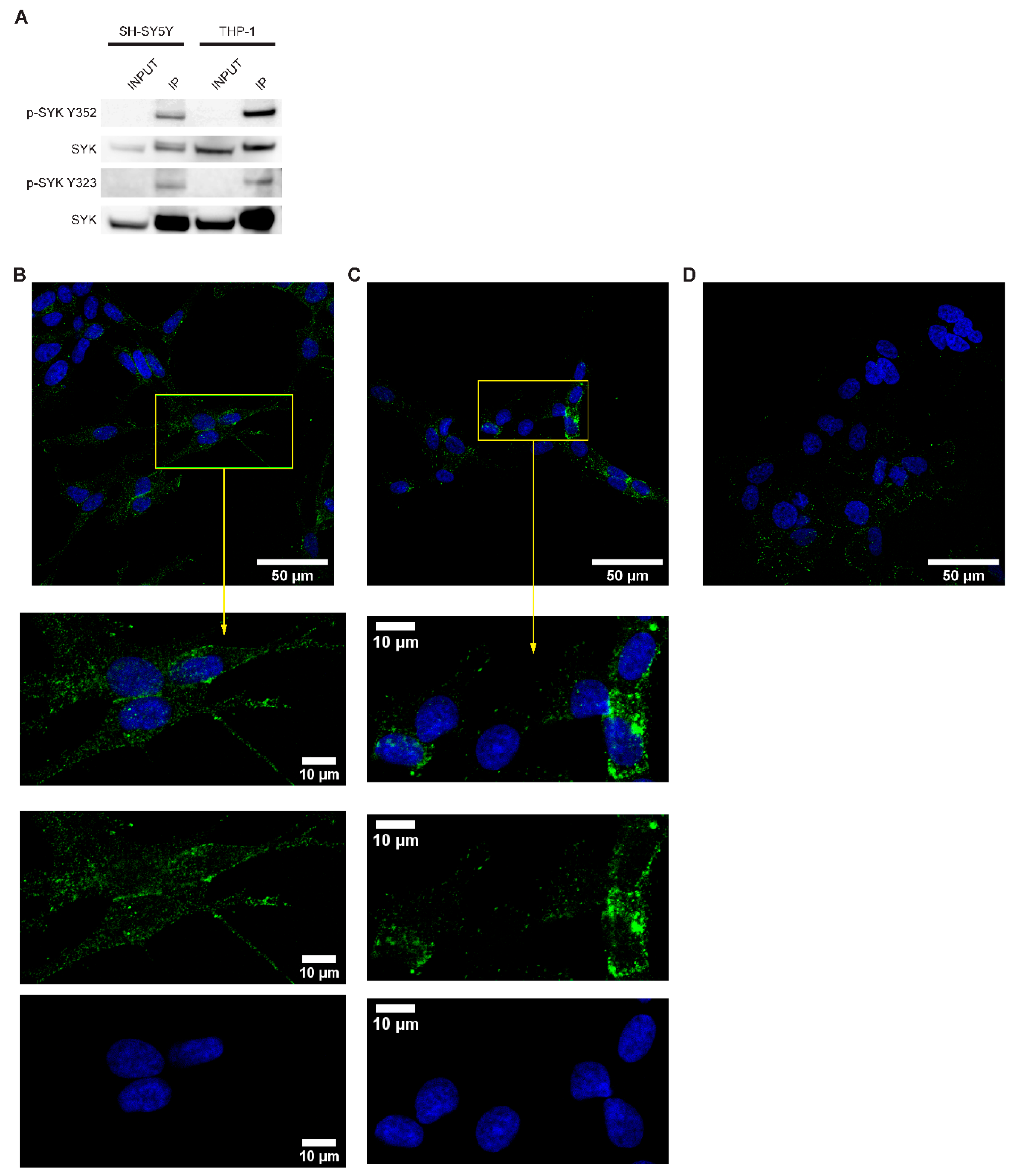
2.3. SYK Is Phosphorylated in Neuroblastoma Cell Lines
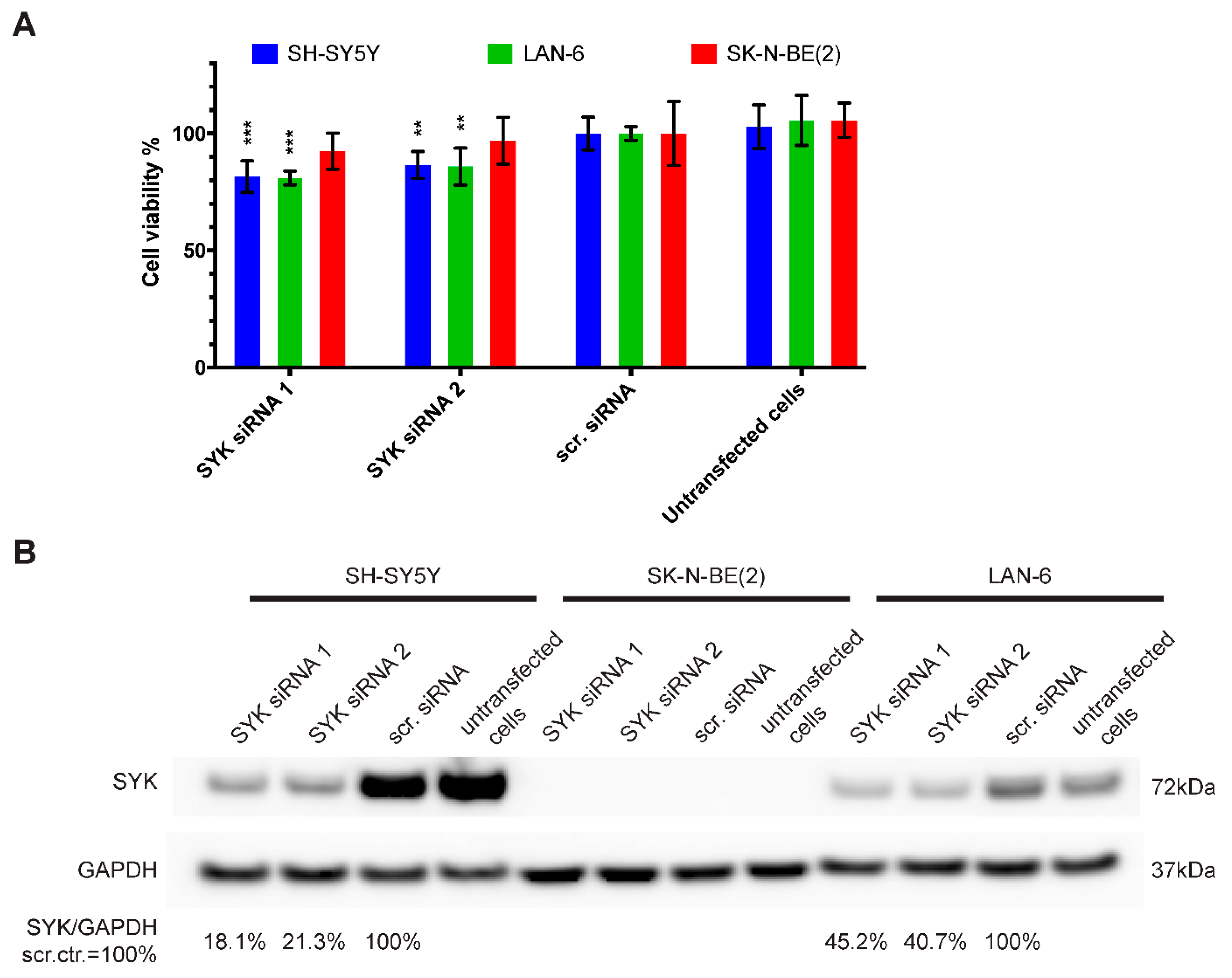
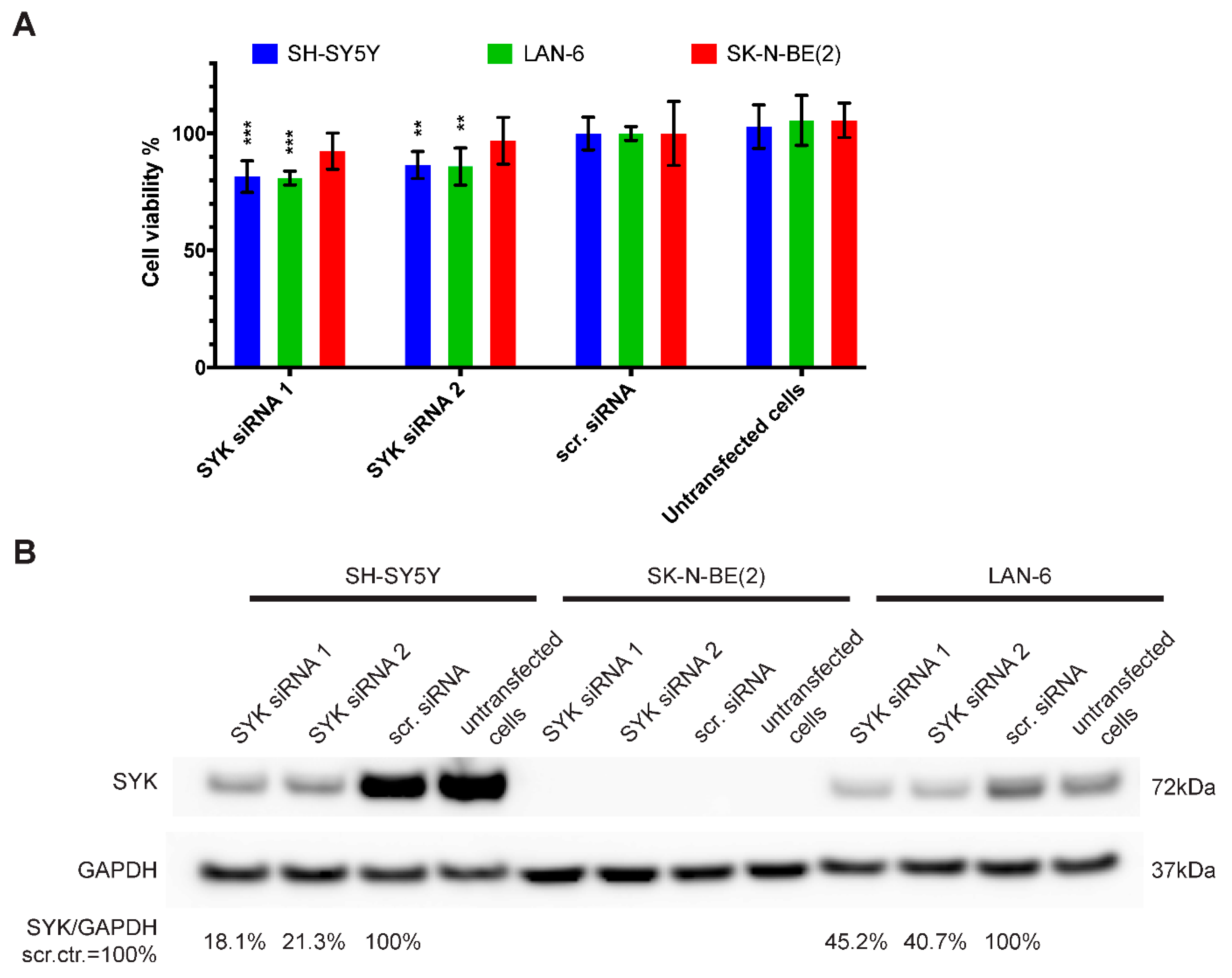
2.4. Downregulation of SYK Reduces the Cell Viability of SYK Expressing Neuroblastoma Cell Lines
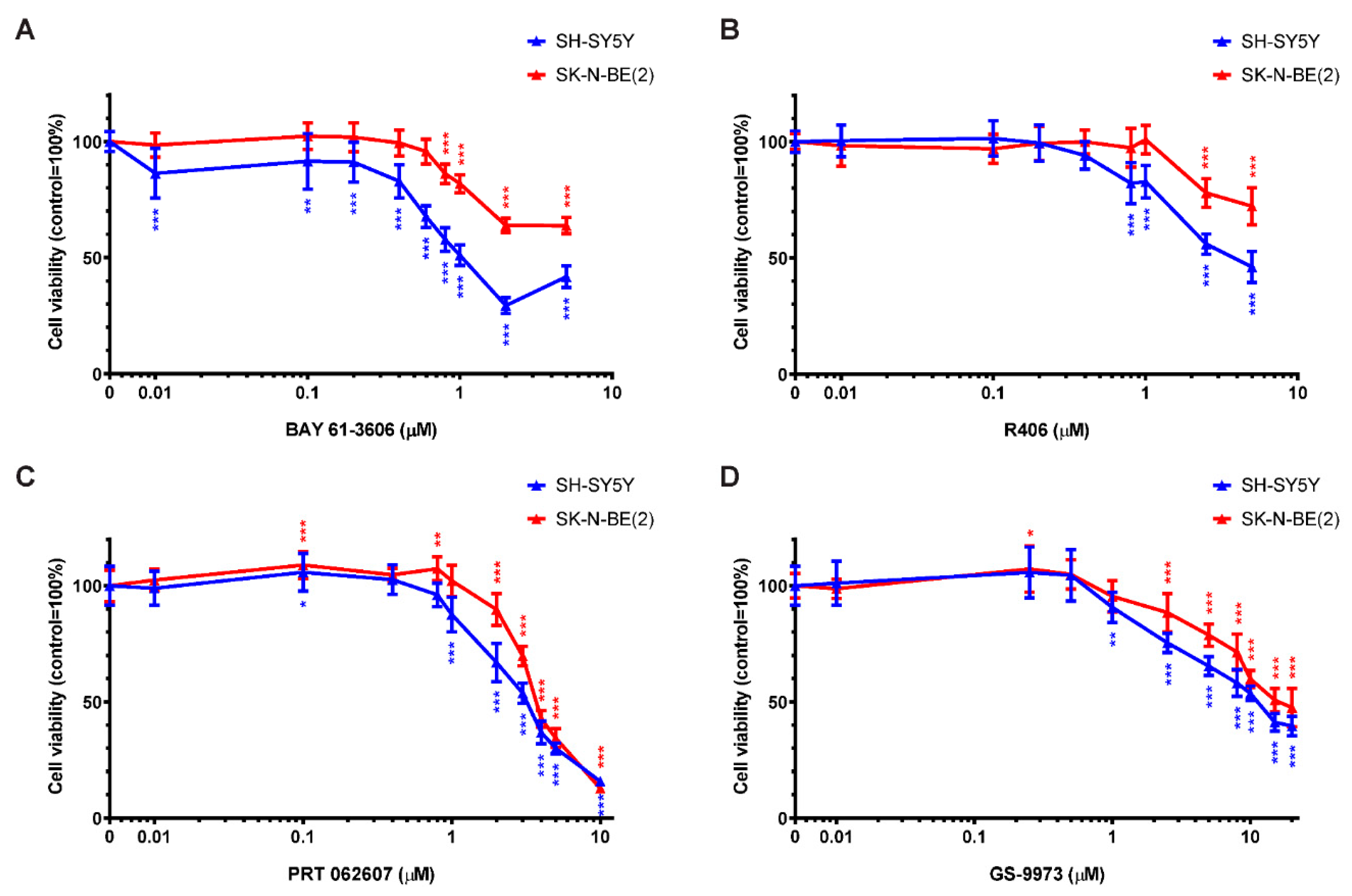
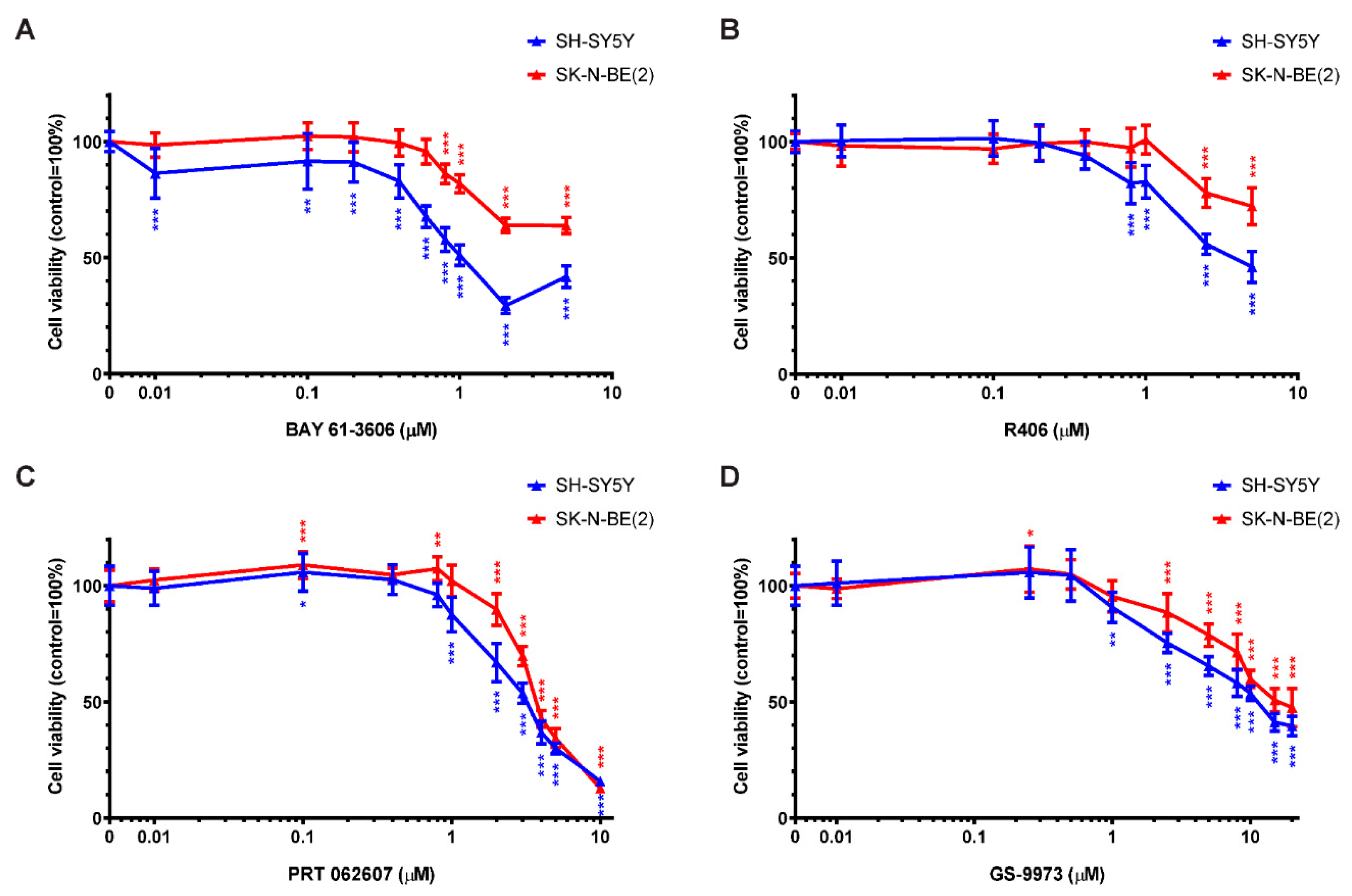
2.5. SYK Activity Inhibition Decreases the Cell Viability of Neuroblastoma Cells
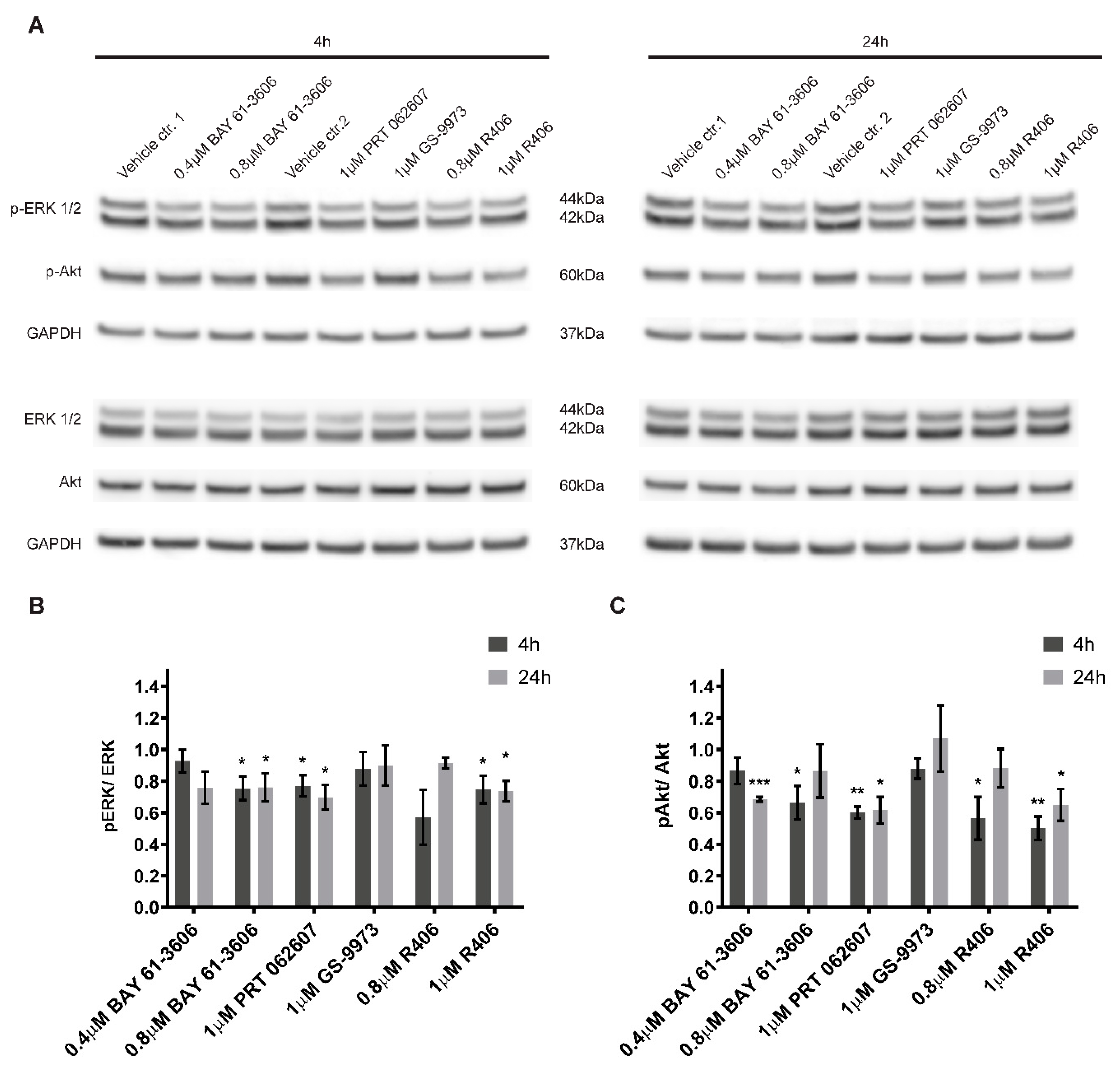
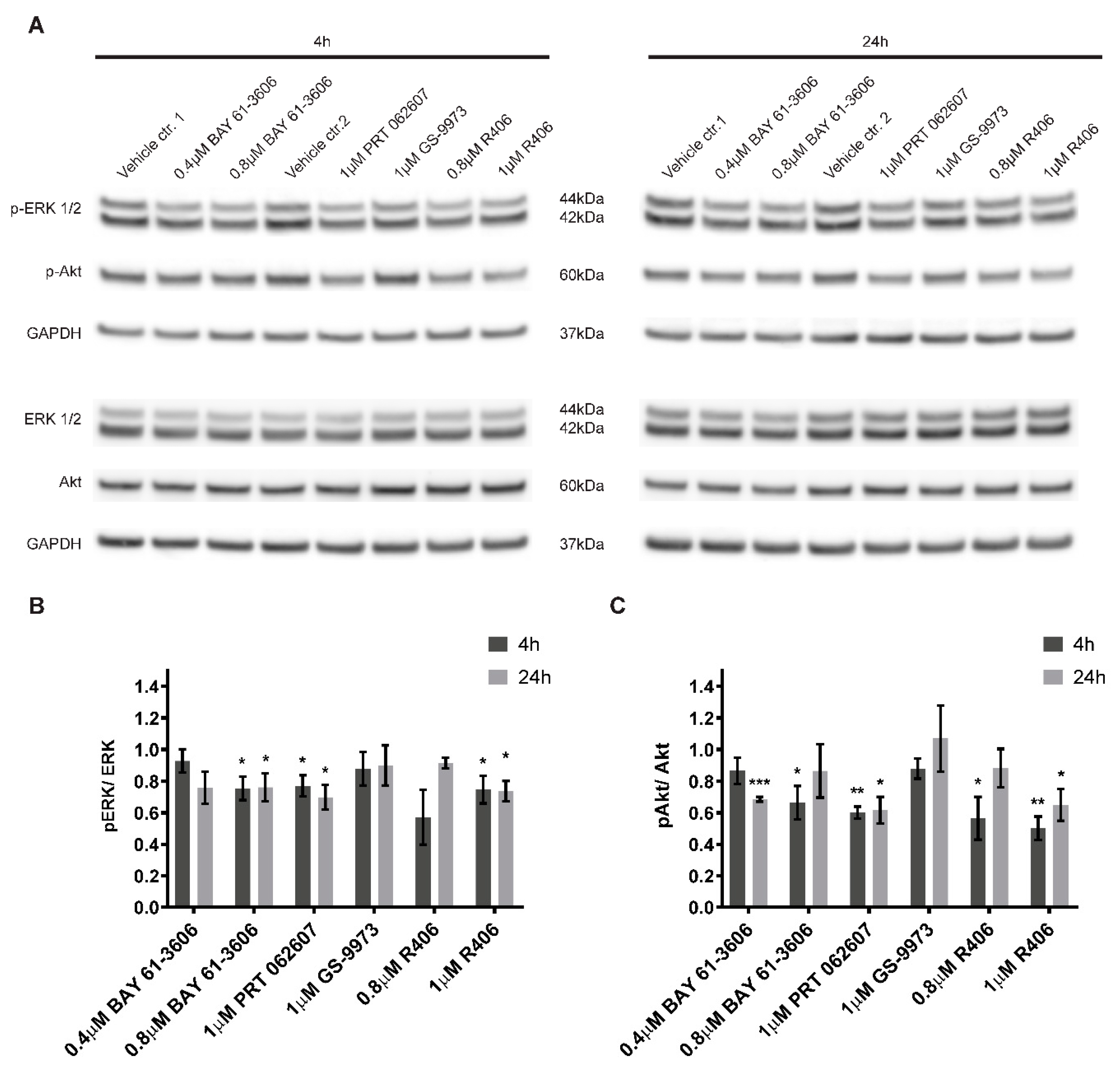
2.6. Inhibition of SYK Activity Reduces ERK1/2 and Akt Phosphorylation in Neuroblastoma Cells
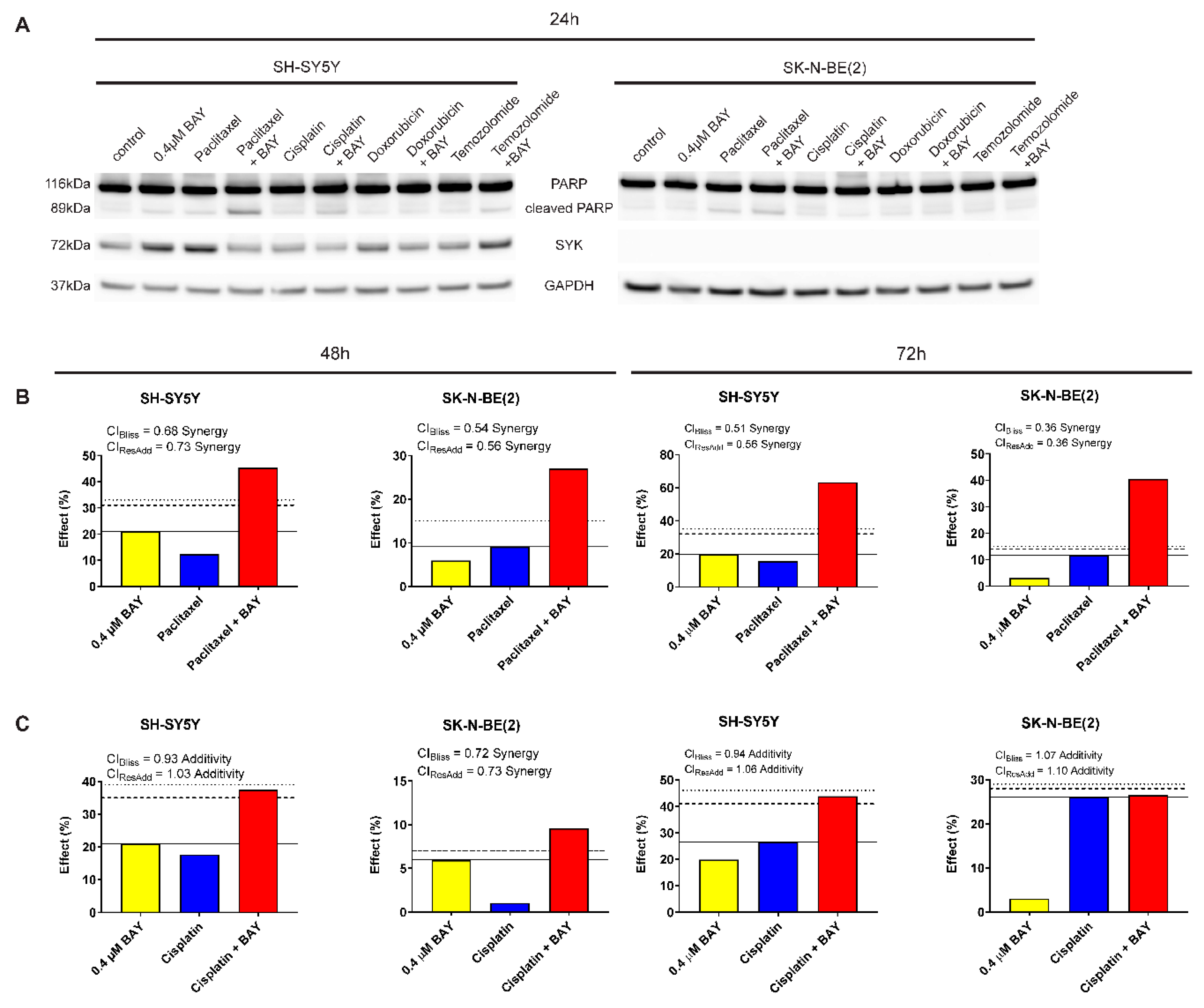
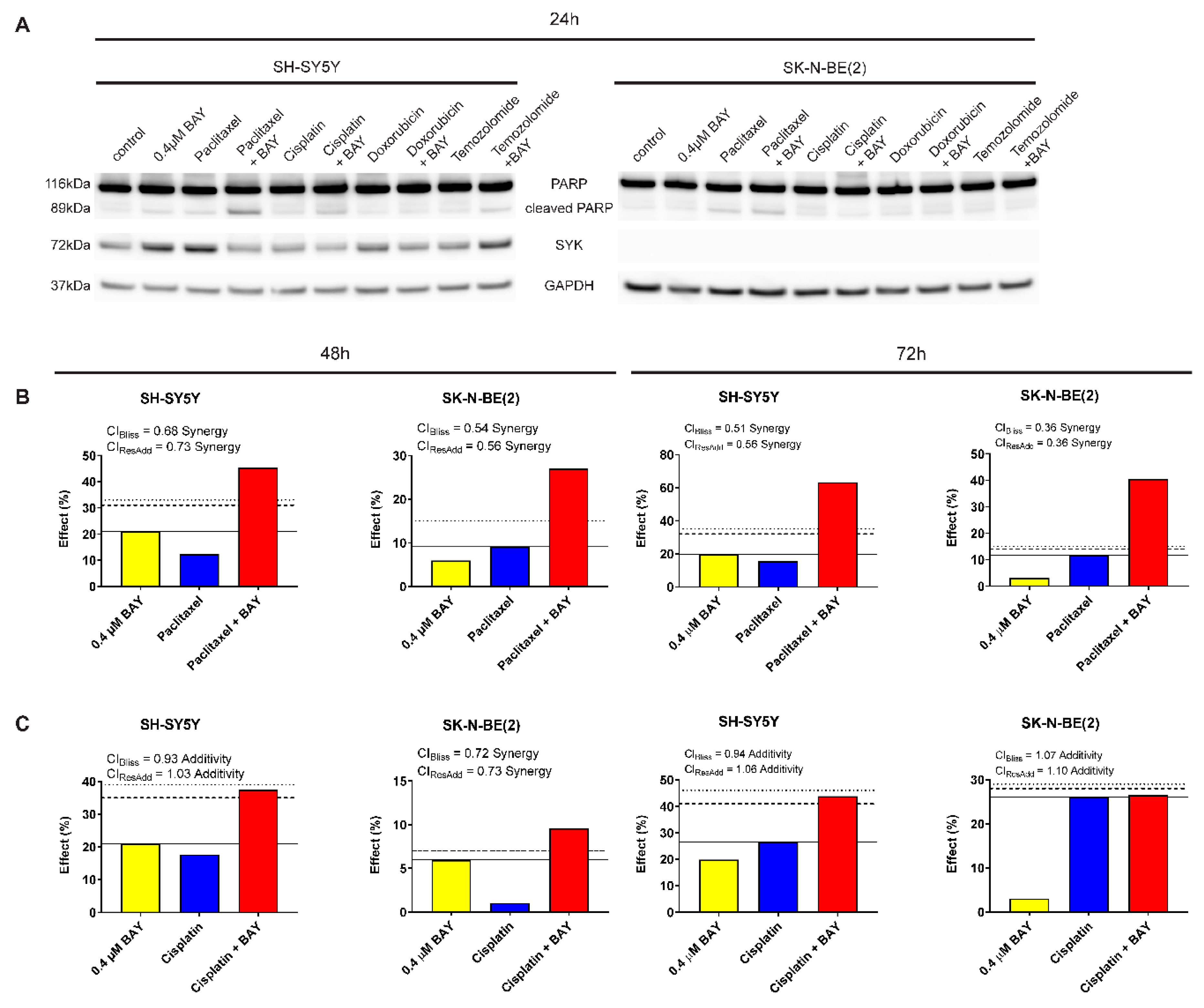
2.7. The Selective SYK Inhibitor BAY 61-3606 Enhances the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells
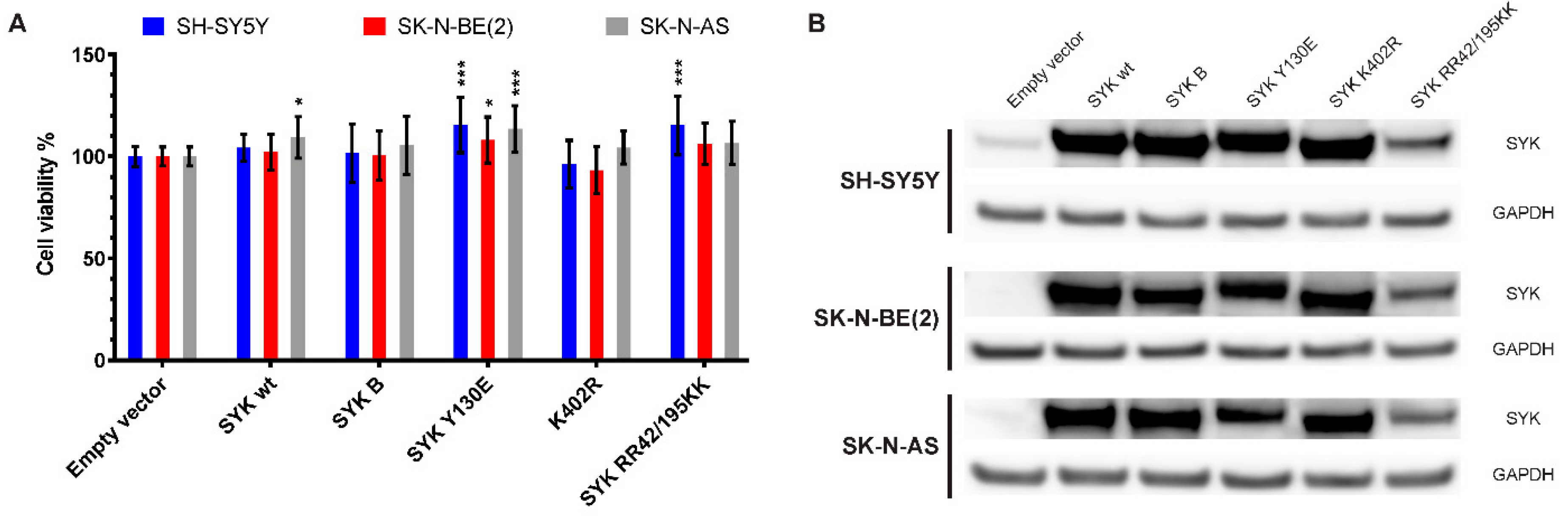
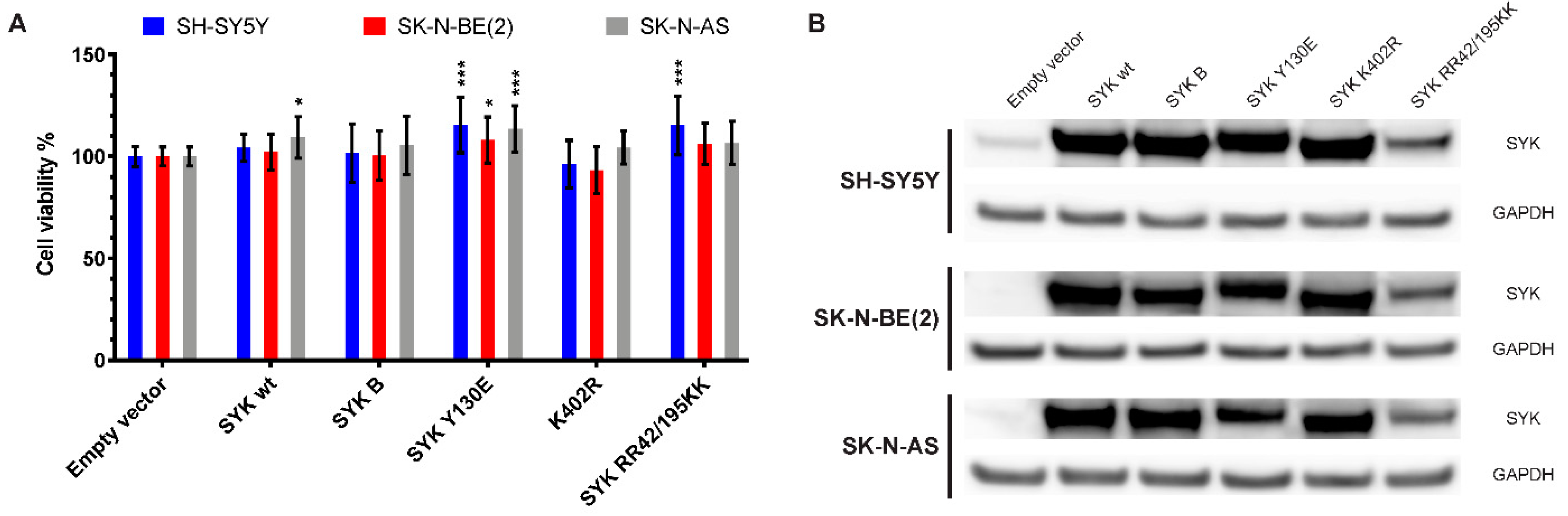
2.8. Transfection with an Active SYK Variant Increases the Cell Viability of Neuroblastoma Cells Independent of Endogenous SYK Levels
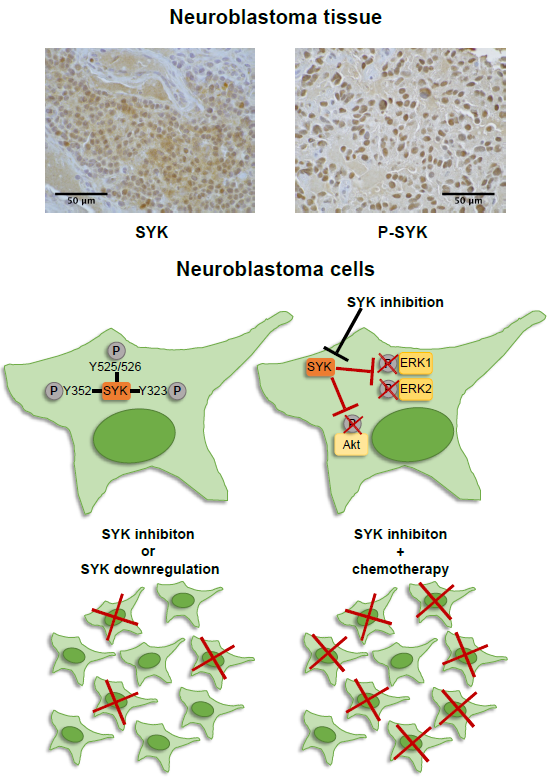
3. Discussion
4. Materials and Methods
4.1. Microarray Gene Expression
4.2. Reagents and Antibodies
4.3. Human Tissue Samples and Cell Lines
4.4. Immunohistochemistry (IHC)
4.5. RNA Isolation and RT-PCR
4.6. Immunocytochemistry (ICC) and Western Blot
4.7. Immunoprecipitation (IP)
4.8. siRNA-Mediated SYK Silencing
4.9. Cell Viability Assay
4.10. Cell Signaling Study
4.11. Transfection with SYK Plasmids
4.12. Drug Combination Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, T. Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Teruko, T.; Alexandra, K. Tyrosine Kinases as Targets for Anti-Inflammatory Therapy. Antiinflam. AntiAllergy Agents Med. Chem. 2007, 6, 47–60. [Google Scholar] [CrossRef]
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Mocsai, A.; Ruland, J.; Tybulewicz, V.L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Yagi, S.; Suzuki, K.; Hasegawa, A.; Okumura, K.; Ra, C. Cloning of the cDNA for the deleted syk kinase homologous to ZAP-70 from human basophilic leukemia cell line (KU812). Biochem. Biophys. Res. Commun. 1994, 200, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Rowley, R.B.; Bolen, J.B.; Fargnoli, J. Molecular cloning of rodent p72Syk. Evidence of alternative mRNA splicing. J. Biol. Chem. 1995, 270, 12659–12664. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Chow, L.M.; Veillette, A. Differential intrinsic enzymatic activity of Syk and Zap-70 protein-tyrosine kinases. J. Biol. Chem. 1996, 271, 22782–22790. [Google Scholar] [CrossRef] [PubMed]
- Geahlen, R.L. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim. Biophys. Acta 2009, 1793, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Mee, P.J.; Costello, P.S.; Williams, O.; Price, A.A.; Duddy, L.P.; Furlong, M.T.; Geahlen, R.L.; Tybulewicz, V.L. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 1995, 378, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Baudot, A.D.; Jeandel, P.Y.; Mouska, X.; Maurer, U.; Tartare-Deckert, S.; Raynaud, S.D.; Cassuto, J.P.; Ticchioni, M.; Deckert, M. The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCdelta and proteasome-dependent regulation of Mcl-1 expression. Oncogene 2009, 28, 3261–3273. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Suryo Rahmanto, Y.; Lee, M.H.; Wu, P.H.; Phillip, J.M.; Huang, C.H.; Vitolo, M.I.; Gaillard, S.; Martin, S.S.; Wirtz, D.; et al. Inhibition of ovarian tumor cell invasiveness by targeting SYK in the tyrosine kinase signaling pathway. Oncogene 2018. [Google Scholar] [CrossRef]
- Angibaud, J.; Louveau, A.; Baudouin, S.J.; Nerriere-Daguin, V.; Evain, S.; Bonnamain, V.; Hulin, P.; Csaba, Z.; Dournaud, P.; Thinard, R.; et al. The immune molecule CD3zeta and its downstream effectors ZAP-70/Syk mediate ephrin signaling in neurons to regulate early neuritogenesis. J. Neurochem. 2011, 119, 708–722. [Google Scholar] [CrossRef]
- Hatterer, E.; Benon, A.; Chounlamountri, N.; Watrin, C.; Angibaud, J.; Jouanneau, E.; Boudin, H.; Honnorat, J.; Pellier-Monnin, V.; Noraz, N. Syk kinase is phosphorylated in specific areas of the developing nervous system. Neurosci. Res. 2011, 70, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Noraz, N.; Jaaoini, I.; Charoy, C.; Watrin, C.; Chounlamountri, N.; Benon, A.; Malleval, C.; Boudin, H.; Honnorat, J.; Castellani, V.; et al. Syk kinases are required for spinal commissural axon repulsion at the midline via the ephrin/Eph pathway. Development 2016, 143, 2183–2193. [Google Scholar] [CrossRef]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd; et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Geahlen, R.L. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 2014, 35, 414–422. [Google Scholar] [CrossRef]
- D’Aura Swanson, C.; Paniagua, R.T.; Lindstrom, T.M.; Robinson, W.H. Tyrosine kinases as targets for the treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 317–324. [Google Scholar] [CrossRef]
- Markham, A. Fostamatinib: First Global Approval. Drugs 2018, 78, 959–963. [Google Scholar] [CrossRef]
- Buchner, M.; Fuchs, S.; Prinz, G.; Pfeifer, D.; Bartholome, K.; Burger, M.; Chevalier, N.; Vallat, L.; Timmer, J.; Gribben, J.G.; et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009, 69, 5424–5432. [Google Scholar] [CrossRef]
- Feng, G.; Wang, X. Role of spleen tyrosine kinase in the pathogenesis of chronic lymphocytic leukemia. Leuk. Lymphoma 2014, 55, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Hardy, I.R.; Clarke, R.L.; Lundy, N.; Pine, P.; Turner, B.C.; Potter, T.A.; Refaeli, Y. Mouse models of non-Hodgkin lymphoma reveal Syk as an important therapeutic target. Blood 2009, 113, 2508–2516. [Google Scholar] [CrossRef]
- Boros, K.; Puissant, A.; Back, M.; Alexe, G.; Bassil, C.F.; Sinha, P.; Tholouli, E.; Stegmaier, K.; Byers, R.J.; Rodig, S.J. Increased SYK activity is associated with unfavorable outcome among patients with acute myeloid leukemia. Oncotarget 2015, 6, 25575–25587. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, S.; Inatome, R.; Takano, T.; Yamamura, H. Syk expression and novel function in a wide variety of tissues. Biochem. Biophys. Res. Commun. 2001, 288, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Krisenko, M.O.; Geahlen, R.L. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 2015, 1853, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Coopman, P.J.; Do, M.T.; Barth, M.; Bowden, E.T.; Hayes, A.J.; Basyuk, E.; Blancato, J.K.; Vezza, P.R.; McLeskey, S.W.; Mangeat, P.H.; et al. The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature 2000, 406, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Layton, T.; Stalens, C.; Gunderson, F.; Goodison, S.; Silletti, S. Syk tyrosine kinase acts as a pancreatic adenocarcinoma tumor suppressor by regulating cellular growth and invasion. Am. J. Pathol. 2009, 175, 2625–2636. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, C.; Thallinger, C.; Pratscher, B.; Bister, M.D.; Schicher, N.; Loewe, R.; Heere-Ress, E.; Roka, F.; Sexl, V.; Pehamberger, H. The non-receptor-associated tyrosine kinase Syk is a regulator of metastatic behavior in human melanoma cells. J. Invest. Dermatol. 2005, 124, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, J.; Li, J.; Wang, L.; Li, M.; Yang, Z.; Zhang, C.; Dai, J.L. Frequent epigenetic inactivation of spleen tyrosine kinase gene in human hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 6687–6695. [Google Scholar] [CrossRef] [PubMed]
- Goodman, P.A.; Wood, C.M.; Vassilev, A.; Mao, C.; Uckun, F.M. Spleen tyrosine kinase (Syk) deficiency in childhood pro-B cell acute lymphoblastic leukemia. Oncogene 2001, 20, 3969–3978. [Google Scholar] [CrossRef]
- Ghotra, V.P.; He, S.; van der Horst, G.; Nijhoff, S.; de Bont, H.; Lekkerkerker, A.; Janssen, R.; Jenster, G.; van Leenders, G.J.; Hoogland, A.M.; et al. SYK is a candidate kinase target for the treatment of advanced prostate cancer. Cancer Res. 2015, 75, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Udyavar, A.R.; Hoeksema, M.D.; Clark, J.E.; Zou, Y.; Tang, Z.; Li, Z.; Li, M.; Chen, H.; Statnikov, A.; Shyr, Y.; et al. Co-expression network analysis identifies Spleen Tyrosine Kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst. Biol. 2013, 7, S1. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gaillard, S.; Phillip, J.M.; Huang, T.C.; Pinto, S.M.; Tessarollo, N.G.; Zhang, Z.; Pandey, A.; Wirtz, D.; Ayhan, A.; et al. Inhibition of Spleen Tyrosine Kinase Potentiates Paclitaxel-Induced Cytotoxicity in Ovarian Cancer Cells by Stabilizing Microtubules. Cancer Cell 2015, 28, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, G.; Grzmil, M.; Smirnova, T.; Zmarz, P.; Huber, R.M.; Hynx, D.; Kohler, H.; Wang, Y.; Hotz, H.R.; Hynes, N.E.; et al. SYK Inhibition Blocks Proliferation and Migration of Glioma Cells, and Modifies the Tumor Microenvironment. Neuro Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, D.C.; Cao, Q.; Pang, B.; Gae, D.D.; Lee, V.K.M.; Lim, H.J.; Doan, N.; Said, J.W.; Gery, S.; et al. Identification of a Novel SYK/c-MYC/MALAT1 Signaling Pathway and Its Potential Therapeutic Value in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 4376–4387. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Duke, L.; Zhang, P.S.; Arlinghaus, R.B.; Symmans, W.F.; Sahin, A.; Mendez, R.; Dai, J.L. Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res. 2003, 63, 4724–4730. [Google Scholar] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; Bouzbid, S.; et al. International incidence of childhood cancer, 2001-10: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Berlanga, P.; Canete, A.; Castel, V. Advances in emerging drugs for the treatment of neuroblastoma. Expert. Opin. Emerg. Drugs 2017, 22, 63–75. [Google Scholar] [CrossRef]
- Johnsen, J.I.; Dyberg, C.; Fransson, S.; Wickstrom, M. Molecular mechanisms and therapeutic targets in neuroblastoma. Pharmacol. Res. 2018, 131, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Billingsley, M.L.; Kincaid, R.L.; Siraganian, R.P. Phosphorylation of Syk activation loop tyrosines is essential for Syk function. An in vivo study using a specific anti-Syk activation loop phosphotyrosine antibody. J. Biol. Chem. 2000, 275, 35442–35447. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.; Jahn, T.; Smith, S.; Lamkin, T.; Uribe, L.; Liu, Y.; Durden, D.L.; Weinberg, K. Differential activation of the tyrosine kinases ZAP-70 and Syk after Fc gamma RI stimulation. Blood 1997, 89, 388–396. [Google Scholar] [PubMed]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Hu, J.; Ma, H.; Harrison, M.L.; Geahlen, R.L. Nucleocytoplasmic trafficking of the Syk protein tyrosine kinase. Mol. Cell. Biol. 2006, 26, 3478–3491. [Google Scholar] [CrossRef] [PubMed]
- Zyss, D.; Montcourrier, P.; Vidal, B.; Anguille, C.; Merezegue, F.; Sahuquet, A.; Mangeat, P.H.; Coopman, P.J. The Syk tyrosine kinase localizes to the centrosomes and negatively affects mitotic progression. Cancer Res. 2005, 65, 10872–10880. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Thompson, D.; Vo, K.T.; London, W.B.; Fischer, M.; Ambros, P.F.; Nakagawara, A.; Brodeur, G.M.; Matthay, K.K.; DuBois, S.G. Identification of patient subgroups with markedly disparate rates of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group project. Cancer 2016, 122, 935–945. [Google Scholar] [CrossRef]
- Wang, L.L.; Teshiba, R.; Ikegaki, N.; Tang, X.X.; Naranjo, A.; London, W.B.; Hogarty, M.D.; Gastier-Foster, J.M.; Look, A.T.; Park, J.R.; et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children’s Oncology Group study. Br. J. Cancer 2015, 113, 57–63. [Google Scholar] [CrossRef]
- Breit, S.; Schwab, M. Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J. Neurosci. Res. 1989, 24, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Nakagawa, Y.; Kawahara, A.; Miyazaki, T.; Sada, K.; Yamamura, H.; Taniguchi, T. Protein tyrosine kinase Syk is associated with and activated by the IL-2 receptor: Possible link with the c-myc induction pathway. Immunity 1995, 2, 89–100. [Google Scholar] [CrossRef]
- Alaminos, M.; Davalos, V.; Cheung, N.K.; Gerald, W.L.; Esteller, M. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J. Nat. Cancer Inst. 2004, 96, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Margetts, C.D.; Morris, M.; Astuti, D.; Gentle, D.C.; Cascon, A.; McRonald, F.E.; Catchpoole, D.; Robledo, M.; Neumann, H.P.; Latif, F.; et al. Evaluation of a functional epigenetic approach to identify promoter region methylation in phaeochromocytoma and neuroblastoma. Endocr. Relat. Cancer 2008, 15, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Grau, E.; Martinez, F.; Orellana, C.; Canete, A.; Yanez, Y.; Oltra, S.; Noguera, R.; Hernandez, M.; Bermudez, J.D.; Castel, V. Epigenetic alterations in disseminated neuroblastoma tumour cells: influence of TMS1 gene hypermethylation in relapse risk in NB patients. J. Cancer Res. Clin. Oncol. 2010, 136, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Grau, E.; Martinez, F.; Orellana, C.; Canete, A.; Yanez, Y.; Oltra, S.; Noguera, R.; Hernandez, M.; Bermudez, J.D.; Castel, V. Hypermethylation of apoptotic genes as independent prognostic factor in neuroblastoma disease. Mol. Carcinog. 2011, 50, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Cottat, M.; Yasukuni, R.; Homma, Y.; Lidgi-Guigui, N.; Varin-Blank, N.; Lamy de la Chapelle, M.; Le Roy, C. Phosphorylation impact on Spleen Tyrosine kinase conformation by Surface Enhanced Raman Spectroscopy. Sci. Rep. 2017, 7, 39766. [Google Scholar] [CrossRef]
- Bohnenberger, H.; Oellerich, T.; Engelke, M.; Hsiao, H.-H.; Urlaub, H.; Wienands, J. Complex phosphorylation dynamics control the composition of the Syk interactome in B cells. European J. Immunol. 2011, 41, 1550–1562. [Google Scholar] [CrossRef]
- Law, C.L.; Chandran, K.A.; Sidorenko, S.P.; Clark, E.A. Phospholipase C-gamma1 interacts with conserved phosphotyrosyl residues in the linker region of Syk and is a substrate for Syk. Mol. Cell. Biol. 1996, 16, 1305–1315. [Google Scholar] [CrossRef]
- Deckert, M.; Tartare-Deckert, S.; Couture, C.; Mustelin, T.; Altman, A. Functional and physical interactions of Syk family kinases with the Vav proto-oncogene product. Immunity 1996, 5, 591–604. [Google Scholar] [CrossRef]
- Groesch, T.D.; Zhou, F.; Mattila, S.; Geahlen, R.L.; Post, C.B. Structural basis for the requirement of two phosphotyrosine residues in signaling mediated by Syk tyrosine kinase. J. Mol. Biol. 2006, 356, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Vanes, L.; Geahlen, R.L.; Tybulewicz, V.L. Distinct roles for the linker region tyrosines of Syk in FcepsilonRI signaling in primary mast cells. J. Biol. Chem. 2005, 280, 4510–4517. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, L.; Laurenti, L.; Gobessi, S.; Longo, P.G.; Leone, G.; Efremov, D.G. Phosphorylation of the activation loop tyrosines is required for sustained Syk signaling and growth factor-independent B-cell proliferation. Cell. Signal. 2009, 21, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kimura, T.; Siraganian, R.P. Mutations in the activation loop tyrosines of protein tyrosine kinase Syk abrogate intracellular signaling but not kinase activity. J. Immunol. 1998, 161, 4366–4374. [Google Scholar] [PubMed]
- Papp, E.; Tse, J.K.; Ho, H.; Wang, S.; Shaw, D.; Lee, S.; Barnett, J.; Swinney, D.C.; Bradshaw, J.M. Steady state kinetics of spleen tyrosine kinase investigated by a real time fluorescence assay. Biochemistry 2007, 46, 15103–15114. [Google Scholar] [CrossRef]
- Prinos, P.; Garneau, D.; Lucier, J.F.; Gendron, D.; Couture, S.; Boivin, M.; Brosseau, J.P.; Lapointe, E.; Thibault, P.; Durand, M.; et al. Alternative splicing of SYK regulates mitosis and cell survival. Nat. Struct. Mol. Biol 2011, 18, 673–679. [Google Scholar] [CrossRef]
- Hong, J.; Yuan, Y.; Wang, J.; Liao, Y.; Zou, R.; Zhu, C.; Li, B.; Liang, Y.; Huang, P.; Wang, Z.; et al. Expression of variant isoforms of the tyrosine kinase SYK determines the prognosis of hepatocellular carcinoma. Cancer Res. 2014, 74, 1845–1856. [Google Scholar] [CrossRef]
- Yamamoto, N.; Takeshita, K.; Shichijo, M.; Kokubo, T.; Sato, M.; Nakashima, K.; Ishimori, M.; Nagai, H.; Li, Y.F.; Yura, T.; et al. The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo [1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. J. Pharmacol. Exp. Ther. 2003, 306, 1174–1181. [Google Scholar] [CrossRef]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K.; Herlaar, E.; Lau, A.; Young, C.; et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 2006, 319, 998–1008. [Google Scholar] [CrossRef]
- Coffey, G.; DeGuzman, F.; Inagaki, M.; Pak, Y.; Delaney, S.M.; Ives, D.; Betz, A.; Jia, Z.J.; Pandey, A.; Baker, D.; et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2012, 340, 350–359. [Google Scholar] [CrossRef]
- Currie, K.S.; Kropf, J.E.; Lee, T.; Blomgren, P.; Xu, J.; Zhao, Z.; Gallion, S.; Whitney, J.A.; Maclin, D.; Lansdon, E.B.; et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J. Med. Chem. 2014, 57, 3856–3873. [Google Scholar] [CrossRef]
- Lin, Y.C.; Huang, D.Y.; Chu, C.L.; Lin, W.W. Anti-inflammatory actions of Syk inhibitors in macrophages involve non-specific inhibition of toll-like receptors-mediated JNK signaling pathway. Mol. Immunol. 2010, 47, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Fey, D.; Halasz, M.; Dreidax, D.; Kennedy, S.P.; Hastings, J.F.; Rauch, N.; Munoz, A.G.; Pilkington, R.; Fischer, M.; Westermann, F.; et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci. Signal. 2015, 8, ra130. [Google Scholar] [CrossRef] [PubMed]
- Colado, A.; Almejún, M.B.; Podaza, E.; Risnik, D.; Stanganelli, C.; Elías, E.E.; Dos Santos, P.; Slavutsky, I.; Fernández Grecco, H.; Cabrejo, M.; et al. The kinase inhibitors R406 and GS-9973 impair T cell functions and macrophage-mediated anti-tumor activity of rituximab in chronic lymphocytic leukemia patients. Cancer Immunol. Immunother. 2016, 66, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ruan, Y.; Tippett, T.; Narendran, A. Targeted inhibition of MEK1 by cobimetinib leads to differentiation and apoptosis in neuroblastoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 104. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.C.; Chockalingam, S.; Melegh, Z.; Greenhough, A.; Malik, S.; Szemes, M.; Park, J.H.; Kaidi, A.; Zhou, L.; Catchpoole, D.; et al. LGR5 regulates pro-survival MEK/ERK and proliferative Wnt/beta-catenin signalling in neuroblastoma. Oncotarget 2015, 6, 40053–40067. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Segerstrom, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kagedal, B.; Eksborg, S.; Sveinbjornsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef]
- King, D.; Yeomanson, D.; Bryant, H.E. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol. Oncol. 2015, 37, 245–251. [Google Scholar] [CrossRef]
- Segerstrom, L.; Baryawno, N.; Sveinbjornsson, B.; Wickstrom, M.; Elfman, L.; Kogner, P.; Johnsen, J.I. Effects of small molecule inhibitors of PI3K/Akt/mTOR signaling on neuroblastoma growth in vitro and in vivo. Int. J. Cancer 2011, 129, 2958–2965. [Google Scholar] [CrossRef]
- Figenschau, Y.; Knutsen, G.; Shahazeydi, S.; Johansen, O.; Sveinbjornsson, B. Human articular chondrocytes express functional leptin receptors. Biochem. Biophys. Res. Commun. 2001, 287, 190–197. [Google Scholar] [CrossRef]
- Tümmler, C.; Snapkov, I.; Wickstrom, M.; Moens, U.; Ljungblad, L.; Maria Elfman, L.H.; Winberg, J.O.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. Inhibition of chemerin/CMKLR1 axis in neuroblastoma cells reduces clonogenicity and cell viability in vitro and impairs tumor growth in vivo. Oncotarget 2017, 8, 95135–95151. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuroblastoma Subgroups | SYK Positive (Sections Examined) | p-SYK Positive (Sections Examined) |
|---|---|---|
| Neuroblastoma | 40 (42) | 38 (40) |
| Non-MYCN-amplified | 31 (32) | 29 (31) |
| MYCN amplified | 9 (10) | 9 (9) |
| * Treated tissue | 11 (13) | 10 (11) |
| * Untreated tissue | 26 (26) | 25 (26) |
| Ganglioneuroma | 3 (3) | 3 (3) |
| SH-SY5Y | SK-N-BE(2) | ||||||
|---|---|---|---|---|---|---|---|
| Treatment | Cell viability (%) Mean ± SD | p value Drug vs. combination | p value BAY vs. combination | Cell viability (%) Mean ± SD | p value Drug vs. combination | p value BAY vs. combination | |
| 48 h | 0.4 μM BAY | 79.01 ± 6.26 | 94.04 ± 7.72 | ||||
| Paclitaxel | 87.71 ± 7.83 | 90.89 ± 7.86 | |||||
| Paclitaxel + BAY | 54.65 ± 3.26 | <0.001 | <0.001 | 72.98 ± 9.33 | <0.001 | <0.001 | |
| Cisplatin | 82.33 ± 9.01 | 98.94 ± 6.48 | |||||
| Cisplatin + BAY | 62.47 ± 5.82 | <0.001 | <0.001 | 90.49 ± 7.46 | 0.052 | >0.999 | |
| Doxorubicin | 100.5 ± 9.59 | 102 ± 9.68 | |||||
| Doxorubicin + BAY | 77.97 ± 8.01 | <0.001 | >0.999 | 93.69 ± 8.78 | 0.121 | >0.999 | |
| Temozolomide | 107 ± 11.17 | 107.7 ± 3.65 | |||||
| Temozolomide + BAY | 76.81 ± 4.82 | <0.001 | >0.999 | 100 ± 4.36 | 0.132 | 0.181 | |
| 72 h | 0.4 μM BAY | 80.20 ± 7.62 | 96.97 ± 6.89 | ||||
| Paclitaxel | 84.53 ± 4.60 | 88.29 ± 5.19 | |||||
| Paclitaxel + BAY | 36.79 ± 3.14 | <0.001 | <0.001 | 59.48 ± 8.81 | <0.001 | <0.001 | |
| Cisplatin | 73.38 ± 5.89 | 73.99 ± 2.95 | |||||
| Cisplatin + BAY | 56.25 ± 5.72 | <0.001 | <0.001 | 73.53 ± 5.94 | >0.999 | <0.001 | |
| Doxorubicin | 102.2 ± 8.37 | 101.3 ± 7.56 | |||||
| Doxorubicin + BAY | 78.57 ± 7.16 | <0.001 | >0.999 | 93.99 ± 9.68 | 0.053 | >0.999 | |
| Temozolomide | 107.7 ± 6.13 | 102.7 ± 4.28 | |||||
| Temozolomide + BAY | 74.21 ± 5.99 | <0.001 | 0.217 | 99.74 ± 6.68 | >0.999 | >0.999 | |
| Antibody | Application | Source |
|---|---|---|
| Anti-SYK | WB, IP | #1240, Santa Cruz Biotechnology |
| Anti-SYK | WB | #13198, Cell Signaling Technology |
| Anti-SYK | ICC/IHC | #HPA001384, Sigma |
| Anti-Phospho-ZAP-70 (Tyr319)/SYK (Tyr352) | WB | #2701, Cell Signaling Technology |
| Anti-Phospho-SYK (Tyr323) | WB | #2715, Cell Signaling Technology |
| Anti-Phospho-SYK (Tyr525/526) | ICC | #2710, Cell Signaling Technology |
| Anti-Phospho-SYK (pTyr525) | IHC | #SAB4503839, Sigma |
| Anti-PARP | WB | #9542, Cell Signaling Technology |
| Anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | WB | #4370, Cell Signaling Technology |
| Anti-p44/42 MAPK (Erk1/2) | WB | #4695, Cell Signaling Technology |
| Anti-Phospho-Akt (Ser473) (D9E) | WB | #4060, Cell Signaling Technology |
| Anti-Akt | WB | #9272, Cell Signaling Technology |
| Anti-GAPDH | WB | #47724, Santa Cruz Biotechnology |
| Goat Anti-Rabbit IgG H&L (HRP) | WB | #6721, Abcam |
| Rabbit Anti-Mouse IgG H&L (HRP) | WB | #97046, Abcam |
| Goat anti-Rabbit IgG (H+L), Alexa Fluor 488 | ICC | # A-11008, Thermo Fisher Scientific |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tümmler, C.; Dumitriu, G.; Wickström, M.; Coopman, P.; Valkov, A.; Kogner, P.; Johnsen, J.I.; Moens, U.; Sveinbjörnsson, B. SYK Inhibition Potentiates the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells In Vitro. Cancers 2019, 11, 202. https://doi.org/10.3390/cancers11020202
Tümmler C, Dumitriu G, Wickström M, Coopman P, Valkov A, Kogner P, Johnsen JI, Moens U, Sveinbjörnsson B. SYK Inhibition Potentiates the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells In Vitro. Cancers. 2019; 11(2):202. https://doi.org/10.3390/cancers11020202
Chicago/Turabian StyleTümmler, Conny, Gianina Dumitriu, Malin Wickström, Peter Coopman, Andrey Valkov, Per Kogner, John Inge Johnsen, Ugo Moens, and Baldur Sveinbjörnsson. 2019. "SYK Inhibition Potentiates the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells In Vitro" Cancers 11, no. 2: 202. https://doi.org/10.3390/cancers11020202
APA StyleTümmler, C., Dumitriu, G., Wickström, M., Coopman, P., Valkov, A., Kogner, P., Johnsen, J. I., Moens, U., & Sveinbjörnsson, B. (2019). SYK Inhibition Potentiates the Effect of Chemotherapeutic Drugs on Neuroblastoma Cells In Vitro. Cancers, 11(2), 202. https://doi.org/10.3390/cancers11020202